|


 Curently online :
11 Curently online :
11 |
|
 Total visitors :
7868505 Total visitors :
7868505 |
|
|
Complementation of Yeast Genes with Human Genes as an Experimental Platform for Functional Testing of Human Genetic Variants
Friday, 2015/11/13 | 08:04:33
|
|
Akil Hamza, Erik Tammpere, Megan Kofoed, Christelle Keong, Jennifer Chiang, Guri Giaever, Corey Nislow and Philip Hieter GENETICS November 1, 2015 vol. 201 no. 3 1263-1274 AbstractWhile the pace of discovery of human genetic variants in tumors, patients, and diverse populations has rapidly accelerated, deciphering their functional consequence has become rate-limiting. Using cross-species complementation, model organisms like the budding yeast, Saccharomyces cerevisiae, can be utilized to fill this gap and serve as a platform for testing human genetic variants. To this end, we performed two parallel screens, a one-to-one complementation screen for essential yeast genes implicated in chromosome instability and a pool-to-pool screen that queried all possible essential yeast genes for rescue of lethality by all possible human homologs. Our work identified 65 human cDNAs that can replace the null allele of essential yeast genes, including the nonorthologous pair yRFT1/hSEC61A1. We chose four human cDNAs (hLIG1, hSSRP1, hPPP1CA, and hPPP1CC) for which their yeast gene counterparts function in chromosome stability and assayed in yeast 35 tumor-specific missense mutations for growth defects and sensitivity to DNA-damaging agents. This resulted in a set of human–yeast gene complementation pairs that allow human genetic variants to be readily characterized in yeast, and a prioritized list of somatic mutations that could contribute to chromosome instability in human tumors. These data establish the utility of this cross-species experimental approach.
See: http://www.genetics.org/content/201/3/1263.abstract?etoc
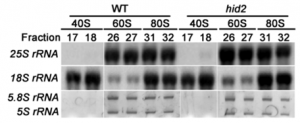
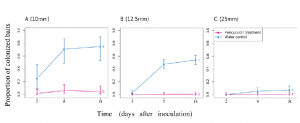
Figure 3 Analyzing features of essential yeast genes that predict replaceability including (A) localization patterns, (B) molecular function, (C) no. of genetic interactions, (D) no. of physical interactions, (E) part of macromolecular complexes, (F) yeast gene size, and (G) human–yeast sequence identity. Localization data, Gene Ontology (GO) terms, no. of genetic/physical interactions, and gene size for each yeast gene were obtained from Yeastmine and each feature is represented as a proportion of the total number of genes input for each set (n = 621 for all essential genes included in both screens and n = 58 for the complementation genes). Overall, the complementation set was enriched for yeast proteins that localize to the cytoplasm (P = 8.18E-03), have catalytic activity (P = 2.28E-02), less physical interactions (P = 5.77E-03), are less likely to be part of macromolecular complexes (P = 3.36E-07), and have smaller gene size (P = 9.16E-03). For sequence identity, “essential gene pairs” refers to the 1076 human–yeast pairs included in this study corresponding to 621 yeast genes and “complementation gene pairs” refers to the 65 complementation pairs corresponding to 58 yeast genes. The box plot highlights the median and range of sequence identity for each set of gene pairs. |
|
|
|
[ Other News ]___________________________________________________
|
(16).png "Complementation of Yeast Genes with Human Genes as an Experimental Platform for Functional Testing of Human Genetic Variants")