|


 Curently online :
7 Curently online :
7 |
|
 Total visitors :
7515475 Total visitors :
7515475 |
|
|
Analysis of tandem gene copies in maize chromosomal regions reconstructed from long sequence reads
Friday, 2016/07/22 | 06:48:50
|
|
Jiaqiang Dong, Yaping Feng, Dibyendu Kumar, Wei Zhang, Tingting Zhu, Ming-Cheng Luo, and Joachim Messing SignificanceGene copy number variation plays an important role in genome evolution and the penetrance of phenotype variations within a species. We have applied new sequencing and physical mapping strategies to obtain long chromosomal regions from a single DNA preparation in each method that comprise tandem repeated gene copies interspersed with transposable elements that comprise about 85% of the genome. This approach should reduce the time and cost to study haplotype variation of complex genomes like those from mammalian and plant species. AbstractHaplotype variation not only involves SNPs but also insertions and deletions, in particular gene copy number variations. However, comparisons of individual genomes have been difficult because traditional sequencing methods give too short reads to unambiguously reconstruct chromosomal regions containing repetitive DNA sequences. An example of such a case is the protein gene family in maize that acts as a sink for reduced nitrogen in the seed. Previously, 41–48 gene copies of the alpha zein gene family that spread over six loci spanning between 30- and 500-kb chromosomal regions have been described in two Iowa Stiff Stalk (SS) inbreds. Analyses of those regions were possible because of overlapping BAC clones, generated by an expensive and labor-intensive approach. Here we used single-molecule real-time (Pacific Biosciences) shotgun sequencing to assemble the six chromosomal regions from the Non-Stiff Stalk maize inbred W22 from a single DNA sequence dataset. To validate the reconstructed regions, we developed an optical map (BioNano genome map; BioNano Genomics) of W22 and found agreement between the two datasets. Using the sequences of full-length cDNAs from W22, we found that the error rate of PacBio sequencing seemed to be less than 0.1% after autocorrection and assembly. Expressed genes, some with premature stop codons, are interspersed with nonexpressed genes, giving rise to genotype-specific expression differences. Alignment of these regions with those from the previous analyzed regions of SS lines exhibits in part dramatic differences between these two heterotic groups.
See: http://www.pnas.org/content/113/29/7949.full PNAS July19 2016; vol.113; no.29: 7949–7956
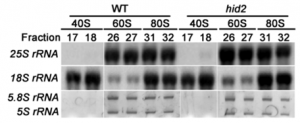
Fig. 3. Alignment of BioNano contigs with assembled PacBio scaffolds. BNG contigs are used as reference (blue bar), with which the scaffolds (green bars) are aligned. The black lines inside green and blues bars are the GCTCTTC sequences recognized by nickase Nt.BspQI. The colored lines on green bars represent supporting contigs for the assemblies. Junctions between colored bars can introduce shifts in the alignments because of gaps in the scaffolds. Contigs are chosen by an empirical confidence score cutoff. For instance, the cyan and yellow contigs contain z1B zein gene copies (third row). Because these contigs are rather short, each of them has a rather low score, and the threshold has been set as 4. However, they are contiguous because both contigs contain z1B zein gene copies in the right order. Therefore, the score of the scaffolds is much higher. |
|
|
|
[ Other News ]___________________________________________________
|
(42).png "Analysis of tandem gene copies in maize chromosomal regions reconstructed from long sequence reads")