|


 Curently online :
25 Curently online :
25 |
|
 Total visitors :
7730555 Total visitors :
7730555 |
|

Fungal plant pathogens are persistent and global food security threats. To invade their hosts they often form highly specialized infection structures, known as appressoria. The cAMP/ PKA- and MAP kinase-signaling cascades have been functionally delineated as positive-acting pathways required for appressorium development. Negative-acting regulatory pathways that block appressorial development are not known.
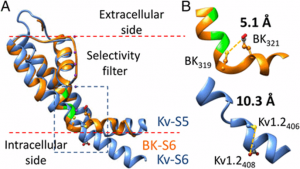
The voltage- and Ca2+-activated BK-type (BK) K+ channels serve as a key regulator of electrical and Ca2+ signaling as well as a model system for studying allosteric regulation of protein. By examining Cd2+ coordination with cysteines placed in BK S6 inner helix, our work reveals a previously undefined structural feature in the BK S6 at a position critical for the coupling between voltage sensor activation and channel opening.

The circadian clock plays a crucial role controlling a wide variety of physiologic and metabolic processes in plants. However, the components and mechanisms involved in such regulation remain to be fully elucidated. Here we show that a protein complex composed of the clock components REVEILLE8/LHY-CCA1-LIKE5 (RVE8/LCL5) and the NIGHT LIGHT–INDUCIBLE AND CLOCK-REGULATED (LNK) proteins has a dual regulatory function.

Detecting quantity trait locus (QTLs) and elite alleles that are associated with grain-filling rate (GFR) in rice is essential for promoting the utilization of hybrid japonica rice and improving rice yield. Ninety-five varieties including 58 landraces and 37 elite varieties from the core germplasm collection were genotyped with 263 simple sequence repeat (SSR) markers. The GFR of the 95 varieties was evaluated at five stages, 7, 14, 21, 28 and 35 days after flowering (DAF) both in 2011 and 2012.

Rice Blast is the most devastating disease causing major yield losses in every year worldwide. It had been proved that using resistant rice varieties would be the most effective way to control this disease. Molecular screening and genetic diversities of major rice blast resistance genes were determined in 192 rice germplasm accessions using simple sequence repeat (SSR) markers. The genetic frequencies of the 10 major rice blast resistance genes varied from 19.79% to 54.69%.

The south of Colombia is a center of diversity for diploid potatoes in the Solanum tuberosum group Phureja. This germplasm is important for genetic studies and is used as a genetic resource in potato breeding programs. In Andean countries, Phureja group potatoes are a staple food and represent important incomes for smallholder farmers.

Plant cells are incapable of sliding past each other, so generation of shape and structure in plant tissue is dependent on cells dividing and expanding in particular directions. Therefore, understanding how cells choose where to build new walls is critical in understanding how plant tissue is patterned. In the present study we expand on previous models of cell division to further understand what parameters of cell geometry and growth influence the position of new walls.

We present a bionic material made of plant cells and carbon nanotubes (CNTs) that exhibits record high temperature sensitivity. This material outperforms by ∼2 orders of magnitude the best man-made materials. The basic mechanism governing this response is the ionic conductivity in the egg-box structure of the pectin backbone, which interconnects cellulose microfibrils in the plant cell wall. The use of CNTs allows the bioelectrical property found in living plants to persist after cell death and stabilizes the response of the dried cells at high temperatures.

One hundred years after its first successful synthesis in the bulk form in 1914, black phosphorus (black P) was recently rediscovered from the perspective of a 2D layered material, attracting tremendous interest from condensed matter physicists, chemists, semiconductor device engineers, and material scientists. Similar to graphite and transition metal dichalcogenides (TMDs)

Agriculture represents the largest water-consuming sector in China, while industry and cities are growing competitors. To sustain a rapidly increasing population with richer diets, high levels of food production have come at significant environmental costs, such as groundwater overdraft and soil degradation. As socioeconomic growth and the associated pressure on water resources continue to increase, it is crucial to evaluate the effects of water-saving measures on agriculture, food trade, and water resources.