|


 Curently online :
6 Curently online :
6 |
|
 Total visitors :
7516099 Total visitors :
7516099 |
|
|
Seed genome hypomethylated regions are enriched in transcription factor genes
Wednesday, 2018/08/29 | 07:49:32
|
|
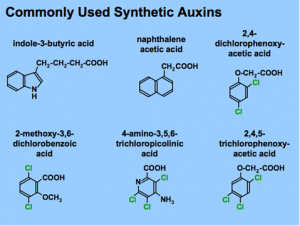
Min Chen, Jer-Young Lin, Jungim Hur, Julie M. Pelletier, Russell Baden, Matteo Pellegrini, John J. Harada, and Robert B. Goldberg PNAS August 28, 2018. 115 (35) E8315-E8322 SignificanceWe scanned soybean and Arabidopsis seed genomes for hypomethylated regions, or DNA methylation valleys (DMVs), present in mammalian cells. Seeds contain DMV regions that have <5% bulk DNA methylation or, in many cases, no detectable DNA methylation. Methylation levels of seed DMVs do not vary detectably during seed development and are present prior to fertilization. Seed DMVs are enriched in transcription factor (TF) genes and are decorated with histone marks that fluctuate developmentally, resembling their animal counterparts in significant ways. We conclude that many genes playing important roles in seed formation are regulated without detectable DNA methylation events and suggest that selective action of TFs, as well as chromatin epigenetic events, play important roles in making a seed. AbstractThe precise mechanisms that control gene activity during seed development remain largely unknown. Previously, we showed that several genes essential for seed development, including those encoding storage proteins, fatty acid biosynthesis enzymes, and transcriptional regulators (e.g., ABI3, FUS3) are located within hypomethylated regions of the soybean genome. These hypomethylated regions are similar to the DNA methylation valleys (DMVs), or canyons, found in mammalian cells. Here, we address the question of the extent to which DMVs are present within seed genomes and what role they might play in seed development. We scanned soybean and Arabidopsis seed genomes from postfertilization through dormancy and germination for regions that contain <5% or <0.4% bulk methylation in CG, CHG, and CHH contexts over all developmental stages. We found that DMVs represent extensive portions of seed genomes, range in size from 5–76 kb, are scattered throughout all chromosomes, and are hypomethylated throughout the plant life cycle. Significantly, DMVs are enriched greatly in transcription factor (TF) genes and other developmental genes that play critical roles in seed formation. Many DMV genes are regulated with respect to seed stage, region, and tissue, and contain H3K4me3, H3K27me3, or bivalent marks that fluctuate during development. Our results indicate that DMVs are a unique regulatory feature of both plant and animal genomes, and that a large number of seed genes are regulated in the absence of methylation changes during development, probably by the action of specific TFs and epigenetic events at the chromatin level.
See: http://www.pnas.org/content/115/35/E8315
Figure 1: Strategy to identify seed DMVs. Seed methylomes (8) were scanned across the genome at each stage of development (arrows) using a 5-kb sliding window with smaller 1-kb incremental steps (Materials and Methods) (dark bracketed lines). The bulk methylation levels in CG, CHH, and CHG contexts were calculated for each window at every developmental stage (8). Genomic regions with bulk methylation levels of <5% or <0.4% across all developmental stages were designated as DMVs, and overlapping DMVs were merged to define DMV regions (red line). |
|
|
|
[ Other News ]___________________________________________________
|
(23).png "Seed genome hypomethylated regions are enriched in transcription factor genes")