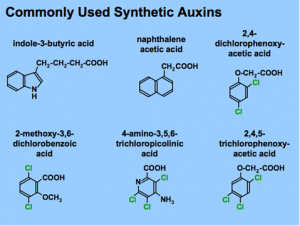
|


 Curently online :
18 Curently online :
18 |
|
 Total visitors :
7731337 Total visitors :
7731337 |
|
|
Recombination in diverse maize is stable, predictable, and associated with genetic load
Sunday, 2015/03/29 | 06:14:42
|
|
Eli Rodgers-Melnicka,1, Peter J. Bradburya,b,1, Robert J. Elshirea, Jeffrey C. Glaubitza, Charlotte B. Acharyaa, Sharon E. Mitchella, Chunhui Lic, Yongxiang Lic, and Edward S. Bucklera,b SignificanceMeiotic recombination is known to vary over 1,000-fold in many eukaryotic organisms, including maize. This regional genomic variation has enormous consequences for plant breeders, who rely on meiotic cross-overs to fine-map quantitative traits and introgress favorable alleles. Deleterious mutations are also predicted to accumulate preferentially within low-recombination regions, particularly within historically outcrossing species, such as maize. Here, we show that meiotic recombination is predictable across diverse crosses based on several genomic features of the reference genome. We demonstrate that the extant patterns of recombination are historically stable and tied to variation in the number of deleterious mutations. The ability of plant breeders to exploit recombination to purge segregating deleterious alleles will determine the efficacy of future crop improvement. AbstractAmong the fundamental evolutionary forces, recombination arguably has the largest impact on the practical work of plant breeders. Varying over 1,000-fold across the maize genome, the local meiotic recombination rate limits the resolving power of quantitative trait mapping and the precision of favorable allele introgression. The consequences of low recombination also theoretically extend to the species-wide scale by decreasing the power of selection relative to genetic drift, and thereby hindering the purging of deleterious mutations. In this study, we used genotyping-by-sequencing (GBS) to identify 136,000 recombination breakpoints at high resolution within US and Chinese maize nested association mapping populations. We find that the pattern of cross-overs is highly predictable on the broad scale, following the distribution of gene density and CpG methylation. Several large inversions also suppress recombination in distinct regions of several families. We also identify recombination hotspots ranging in size from 1 kb to 30 kb. We find these hotspots to be historically stable and, compared with similar regions with low recombination, to have strongly differentiated patterns of DNA methylation and GC content. We also provide evidence for the historical action of GC-biased gene conversion in recombination hotspots. Finally, using genomic evolutionary rate profiling (GERP) to identify putative deleterious polymorphisms, we find evidence for reduced genetic load in hotspot regions, a phenomenon that may have considerable practical importance for breeding programs worldwide.
See http://www.pnas.org/content/112/12/3823.abstract.html?etoc PNAS March 24, 2015 vol. 112 no. 12 3823-3828
Fig. 1. Genome-wide cross-over density in US-NAM and its association with CpG methylation and CN-NAM. Kernel density estimates of cross-over density are shown by both height and color, relative to the maximum density across all chromosomes, and black lines give the relative frequency of methylated CpGs, with scales given on the right side. The locations of centromeres are shown in gray. (Inset) Relationship between 1-Mb cross-over counts of US-NAM and CN-NAM populations is given. |
|
|
|
[ Other News ]___________________________________________________
|
.gif "Recombination in diverse maize is stable, predictable, and associated with genetic load")