- - Genome-scale knockout simulation and clustering analysis of drug-resistant breast cancer cells reveal drug sensitization targets
- - With Science We Can documentary campaign
- - Mapping for Resilience: How Spatial Data is Transforming Karamoja Cluster
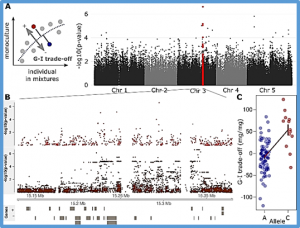
- - Fine-mapping of sm6.1, a novel locus conferring resistance to gray leaf spot from Solanum habrochaites accession LA1777
- - Make forensic case reports required legal and scientific documents
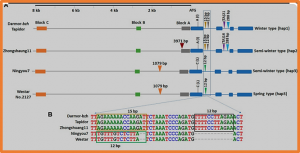
- - German Media Shift Toward a More Favorable Stance on Gene Technology with NGTs
- - Fertile interspecific diploid hybrids between the Asian and African rice species facilitated by tetraploidization and its reduction
- - Review Paper Outlines Vital Role of Regulation in Plant Gene Editing
- - Scientists Use CRISPR to Develop Ideal Plant Architecture Traits in Rice
- - Targeted Editing of the miR529/miR156-IPA1 Regulatory Module via the CRISPR/Cas9 System for Developing
- - Experts from India Use Gene Editing to Fight Rice Dehydration
- - EFSA GMO Panel Releases Scientific Opinion on GM Cotton MON 88913
- - OsDUF2488 acts synergistically with OsPrx1.1, regulates ROS metabolism and promotes dehydration tolerance in rice
- - USask Researchers Discover Pair of Genes that Protect Wheat from Stripe Rust
- - Australian Chickpea Pan-genome to Boost National Chickpea Production
- - Genomic insights into assembly of α-β hydrolase superfamily genes involved in blast resistance in rice
- - COGEM Releases Advice on GM Soybean 305423xDAS-44406-6
- - GM Yeast to Produce Valuable Materials from Urine
- - Heritable variation in root emergence during post-drought recovery reveals potential links to seedling drought recovery in rice
- - Australia`s Gene Technology Regulator Grants Approval for GM Canola Field Trial
- - FSANZ Board Approves Updated Definition of Gene Technology and New Breeding Techniques
- - A distinct LHCI arrangement is recruited to photosystem I in Fe-starved green algae
- - IITA deepens national partnerships for agricultural transformation in Benin
- - A Bioconductor workshop empowers African scientists in genomic data analysis
- - Genetic mapping and validation of QTL for whitefly resistance in cassava (Manihot esculenta Crantz)
- - New study proposes smarter cropland allocation to support sustainable agricultural intensification in Senegal
- - Study Examines Ethical Use of Genetic Modification to Eliminate Harmful Species
- - Sparse testing designs for optimizing resource allocation in multi-environment cassava breeding trials
- - KAIST Researchers Pioneer RNA Modification in Living Organisms
- - EFSA GMO Panel Releases Scientific Opinion on GM Soybean MON87708
- - Anticancer Potential of Pineapple and its Bioactive Compound Bromelain
- - TU Wien Scientists Engineer Bacteria to Metabolize Carbon Monoxide
- - Pairwise Licenses Gene Editing Platform to CIMMYT
- - Homeodomain leucine zipper protein controls the lobed leaf formation by modulating auxin distribution in watermelon
- - Indonesia Showcases Agricultural Biotechnology Innovations to Stakeholders
- - ICRISAT Develops World`s First Extreme Heat-Tolerant Pigeonpea via Speed Breeding
- - Single-Cell and Spatial Transcriptomics Reveals a Stereoscopic Response of Rice Leaf Cells to Magnaporthe oryzae Infection
- - IRRI launches new 5-Year Strategy to drive sustainability in rice-based agri-food systems
- - New Center of Excellence Launched to Drive Agricultural Transformation through South-South Cooperation
- - Integration of BSA-seq and high-resolution mapping reveals genomic regions and candidate genes controlling seed oil accumulation in peanut (Arachis hypogaea L.)
- - Breakthrough at ICRISAT: World’s First Extreme Heat-Tolerant Pigeonpea Developed via Speed Breeding
- - From India to Saudi Arabia and Sudan: Scaling Up Zinc-Enriched Sorghum for Global Impact
- - Degradation of biodegradable plastic films in soil: microplastics formation and soil microbial community dynamics
- - Raising productivity and profits, How AgWise is Closing Yield Gaps through AI
- - Scaling Climate-Smart Water Solutions: IWMI`s Innovation Bundles and Pathways to Impact under CGIAR`s Scaling for Impact Initiative
- - Prospects for cereal self-sufficiency in sub-Saharan Africa
- - A smarter way to grow rice
- - Philippines introduces innovative satellite-based crop insurance and agro-advisory services to help build farmers` resilience
- - Transcriptomic analysis of AhAHL23-mediated root development and space-induced mutations in peanut (Arachis hypogaea L.)
- - Global Agrifood Biotechnologies Conference 2025 opens with a call for inclusive and responsible innovation
- - FAO and WFP early warning report reveals worsening hunger in 13 hotspots; five with immediate risk of starvation
- - Universal features of alternative splicing and the regulatory roles of transcription factors in this process under diverse environmental stimuli in rice
- - FAO at the 2025 United Nations Ocean Conference
- - As famine data dries up, can AI step in? (Devex)
- - ZmPRR37-ZmYSL14 module enhances salt stress tolerance in maize
- - A New Chapter for Goat Breeding in Pakistan
- - What`s really in our food? A global Look at Food Composition Databases—and the Gaps We Need to Fix
- - Ubiquitin Ligase Gene OsPUB57 Negatively Regulates Rice Blast Resistance
- - Regulatory Challenges Slow India`s Progress on GM Crop Adoption
- - USDA-APHIS Designates Cibus`s Herbicide Tolerance Canola Trait HT2 as Not Regulated
- - Improving panicle blast resistance and fragrance in a high-quality japonica rice variety through breeding
- - Australia`s Gene Regulator Approves Field Trial of GM Safflower
- - CAS Experts Optimize Tomato Production for Vertical Farming
- - Control of seed-to-seedling transition by an upstream open reading frame in ABSCISIC ACID DEFICIENT2
- - EPA Announces Approval of GM Cholera Vaccine in New Zealand
- - Experts Urge EU to Allow NGTs in Organic Farming
- - DSD1/ZmICEb regulates stomatal development and drought tolerance in maize
- - CAAS Researchers Examine Factors Affecting GM Maize Adoption in China
- - CSIRO Scientists Identify Gene Behind Bt Cotton Resistance Against Bollworms
- - 110 years of rice breeding at LSU: realized genetic gains and future optimization
- - Australia`s Gene Technology Regulator Approves Field Trial of GM Canola
- - Study Identifies Drivers of Efficient, Precise Genome Editing and Hidden DNA Repair
- - X and Y gene dosage effects are primary contributors to human sexual dimorphism: The case of height
- - Study Identifies Drivers of Efficient, Precise Genome Editing and Hidden DNA Repair
- - China Approves Three GMM-Derived Enzymes
- - A plastid lipid-associated protein-encoding gene (GhPAP) that positively regulates fiber strength was identified via genetic mapping and transcriptomic analysis of a stable QTL on chromosome D06 of upland cotton
- - Kashmir University Produces India`s First Gene-Edited Sheep
- - Save the Date: ASCA8 in Manila - September 2025
- - Genome-Wide Identification of GmARF9b/ GmARF2a Negatively Regulate Root Growth in Soybean
- - Japan and IRRI`s AGRI aims to reduce GHG emissions via CapDev and infrastructure improvements
- - South-South Cooperation: Crucial for Transforming Agriculture
- - Genome-Wide Identification and Expression Analysis of the Mediator Complex Subunit Gene Family in Cassava
- - Telangana`s First Mobile Soil Lab Ignites a Movement for Environmental Wellness
- - Tackling water scarcity requires speed, scale and determination, FAO says
- - Improving plant breeding through AI-supported data integration
- - Astana International Forum: FAO calls for increased investment to unlock Central Asia`s potential and address regional challenges
- - FAO Director-General honours King Abdullah II of Jordan with the prestigious FAO Agricola Medal, praising the monarch`s regional leadership on food security
- - Rhomboid-mediated cleavage of the immune receptor XA21 protects grain set and male fertility in rice
- - Preventing the next pandemic: One Health researcher calls for urgent action
- - New Center of Excellence Launched to Drive Agricultural Transformation through South-South Cooperation
- - tRNA selectivity during ribosome-associated quality control regulates the critical sterility-inducing temperature in two-line hybrid rice
- - Director-General QU Dongyu - Bilateral meeting with the Prime Minister of Ethiopia, H.E. Abiy Ahmed
- - Scientists Review RNAi-induced Effects in GM Plants
- - CAS9 Mediated In-Planta Defence Strategy Against Tomato Leaf Curl New Delhi Virus (ToLCNDV) in Tomato
- - Fewer Off-Target Gene Editors Using AI
- - Scientists Use Gene Editing to Combat Tomato Leaf Curl New Delhi Virus
- - Host-induced gene silencing of the amino acid biosynthesis gene acetolactate synthase of Phytophthora infestans caused strong enhanced late blight resistance of potato in the field
- - GM Rice Shows Enhanced Resistance to Salinity Stress
- - Revised Regulations on the Protection of New Plant Varieties Released in China
- - Resistance Spectrum Analysis and Breeding Utilization of Rice Blast Resistance Gene Pigm-1
- - Kenyan Farmers Seek Financial Assistance in Adopting Bt Cotton
- - Texas A&M AgriLife Scientists Find New Defense Against Hard-To-Treat Plant Diseases
- - The white lupin CCR1 receptor-like kinase controls systemic Autoregulation of Cluster Root and Nodule Development
- - Experts Support Product-Based Regulatory Approach for GMOs
- - EFSA Releases Assessment of GM Oilseed Rape MON 88302
- - Development of Genetically Modified Rice with Enhanced Resistance to Salinity Stress
- - Australian OGTR Receives License Application for Commercial Release of GM Purple Tomato
- - ISAAA Announces New Dates and Venue for ASCA8: Event Relocates to Manila in September 2025
- - The evolutionary history of the common bean (Phaseolus vulgaris) revealed by chloroplast and nuclear genomes analysis
- - What does process improvement look like for CGIAR crop breeding programs?
- - Drylands under pressure: Science and solutions for global stability
- - tRNA selectivity during ribosome-associated quality control regulates the critical sterility-inducing temperature in two-line hybrid rice
- - Director-General QU Dongyu - Bilateral meeting with the Prime Minister of Ethiopia, H.E. Abiy Ahmed
- - Scientists Review RNAi-induced Effects in GM Plants
- - CAS9 Mediated In-Planta Defence Strategy Against Tomato Leaf Curl New Delhi Virus (ToLCNDV) in Tomato
- - Fewer Off-Target Gene Editors Using AI
- - Scientists Use Gene Editing to Combat Tomato Leaf Curl New Delhi Virus
- - Genome-wide association study to identify candidate genes for submergence tolerance during rice seed germination
- - CRISPR Sheds Light on Enhancing Nitrogen Fixation in Bean Genes
- - Scientists Develop Gene Editing Method to Reduce Corn Plant Height
- - Oryza genome evolution through a tetraploid lens
- - OsKCS11 Links Very-Long-Chain Fatty Acids and Cytokinin in Rice Dwarfism
- - Study Reveals Genetic Secrets of Rice
- - The knockout of ClaCSLH1 induced dwarfing in watermelon
- - Infant is World`s First Patient Treated with Customized CRISPR Gene Editing Therapy
- - Gene Editing Offers Dual Protection Against PRV and PRRSV
- - CRISPR-Cas9-mediated knockout of OsKCS11 in rice reveals potential crosstalk between very-long-chain fatty acids and cytokinin
- - Columbia University and Broad Institute Researchers Develop New Gene Editor for Safer, More Precise Gene Therapies
- - UK`s Precision Breeding Act for Plants Signed into La
- - Overexpression of the Transcription Factor GmbZIP60 Increases Salt and Drought Tolerance in Soybean (Glycine max)
- - Kobe University Researchers Produce Healthier Yogurt Using Gene Editing
- - Microbiologist from São Paulo Named 2025 World Food Prize Laureate
- - Directional improvement of agronomic traits in salt-tolerant rice by multiplex-genome-editing
- - Multiplex Genome Editing Improves Agronomic Traits of Salt-tolerant Rice SR86
- - EFSA GMO Panel Releases Scientific Opinion on GM Sugar Beet KWS20-1
- - Optical mapping in plant comparative genomics
- - Ecuador Determines Cibus Herbicide Tolerance Rice Traits are Equivalent to Conventional Breeding
- - Experts Discuss Barriers to Bt Cotton Adoption in the Philippines
- - Chromosome-level genome assembly of a high-yield Chinese soybean variety Mengdou1137 unlocks genetic potential of disease and lodging resistance
- - Gene Editing Spiders Produce Red Fluorescent Silk
- - Save the Date: 8th Asian Short Course on Agribiotechnology, Biosafety Regulation, and Communication
- - Mechanism Analysis of OsbHLH34-OsERF34 Mediated Regulation of Rice Resistance to Sheath Blight
- - Empowering dryland communities through drought early warnings to enhance resilience
- - CGIAR`s digital leap: charting a new course for agriculture through innovation and inclusion
- - Proviral insights of glycolytic enolase in Bamboo mosaic virus replication associated with chloroplasts and mitochondria
- - FAO wins prize for geospatial project in Zimbabwe
- - FAO participates in ADB annual meeting, strengthens cooperation in key agricultural areas
- - QTL mapping and multi-omics identify candidate genes for protein and oil in soybean
- - FAO warns: Enhanced awareness and action needed amid foot-and-mouth disease outbreaks in Europe and the Near East
- - Cooperation at all levels and funding “critical” for plant health and food security, FAO says
- - Identification of peptidome-based biomarkers of cassava mosaic disease resistance in different cassava varieties
- - Researchers Use Salmon DNA to Develop Cold-Tolerant Tilapia
- - Experts from UC San Diego Push Genetically Enhanced Crops for Carbon Dioxide Removal
- - Status on Genetic Resistance to Rice Blast Disease in the Post-Genomic Era
- - 27 EU Agri-food Value Chain Partners Sign Joint Position Paper on NGT Traceability and Labelling
- - Gene-edited Rice Provides Insights into Abiotic Stress Resistance
- - A rare stop-gain SNP mutation in BrGL2 causes aborted trichome development in Chinese cabbage (Brassica rapa L. ssp. pekinensis)
- - UC Davis Develops Wheat with Reduced Gluten Proteins
- - Gene-Edited PRRS-Resistant Pigs Get US FDA Approval
- - A Knockout of the OsGAPDHC6 Gene Encoding a Cytosolic Glyceraldehyde-3-Phosphate Dehydrogenase Reacts Sensitively to Abiotic Stress in Rice
- - India Releases Two Genome-Edited Rice Varieties
- - Save the Date: 8th Asian Short Course on Agribiotechnology, Biosafety Regulation, and Communication
- - Allele-based modeling to predict phenological stages of grapevine hybrids under future climatic conditions
- - India Releases Two Genome-Edited Rice Varieties
- - Save the Date: 8th Asian Short Course on Agribiotechnology, Biosafety Regulation, and Communication
- - Discovery of the widespread site-specific single-stranded nuclease family Ssn
- - Enhancing Transformation of South Korean Maize Cultivar with CRISPR
- - Gene Editing Improves Oleic Acid and Stabilizes Bran Oil in Rice
- - Efficient breeding of high oleic rice cultivar by editing OsFAD2-1 via CRISPR/Cas9
- - INRS Researchers Discover New Tool for Cutting Single-stranded DNA
- - UCLA and UC Berkeley Scientists Develop Tiny CRISPR Tool for Faster, Simpler Plant Genome Editing
- - Australia-backed project aimed to improve cassava genetics in Vietnam
- - Map-based cloning of Zmccr3 and its network construction and validation for regulating maize seed germination
- - New CRISPR Technique Reveals Hidden Microbial Diversity
- - Pangenome Analysis Reveals Genetic Key to Larger Peanut Yields
- - Transgenic cowpea conferring insect resistance and glyphosate tolerance
- - Study Unravels Cause of Tomato Bacterial Spot`s Global Spread
- - Analysis Identifies Specific Genes Found in All Grasses
- - Harnessing neo-domestication of wild pigmented rice for enhanced nutrition and sustainable agriculture
- - Transgenic Cowpea Confers Insect Resistance and Herbicide Tolerance
- - USDA-APHIS Designates Cibus` Disease Resistance Traits for Gene-Edited Canola as Not Regulated
- - Genome-wide association study to identify candidate genes for submergence tolerance during rice seed germination
- - CRISPR Sheds Light on Enhancing Nitrogen Fixation in Bean Genes
- - Scientists Develop Gene Editing Method to Reduce Corn Plant Height
- - Research advances in rice blast resistance genes
- - From data to impact: IRRI`s digital vision at CGIAR Science Week 2025
- - Rising from Drought: Resilient Harvests Flourish in the Sahel
- - GCN5-related histone acetyltransferase HOOKLESS2 regulates fungal resistance and growth in tomato
- - ICRISAT celebrates World IP Day, forging new partnership with BITS Pilani
- - IRRI and ICRISAT Set a Joint Vision to demonstrate Integrated Seed Systems for Dryland Farming in South Asia
- - Combined analysis of transcriptomics and metabolomics showed that SNAC4 and SNAC9 are negative regulators of the resistance to Botrytis cinerea in tomato
- - ICRISAT and SERP Join Hands to Revitalize Livelihoods in Telangana through Agro-Processing and Entrepreneurship Development
- - Boost for the global fight against illegal, unreported and unregulated fishing
- - Functional redundancy in the toxic pathway of Bt protein Cry1Ab, but not Cry1Fa, against the Asian corn borer
- - FAO launches a $150 million Emergency and Early Recovery Response Plan for Ukraine to support war-affected rural communities
- - FAO launches AIM4NatuRe to improve monitoring of ecosystem restoration
- - Diet-regulated transcriptional plasticity of plant parasites in plant–mutualist environments
- - In Kyrgyzstan, Director-General plants seedlings, opens photo exhibit and visits agricultural facilities
- - Launch webinar: toolkit for anticipatory action in fragile, conflict- and violence-affected settings
- - OsBSK3 and OsBSK2 regulate grain size and leaf angle via MAPK signaling pathway in rice
- - The Power of Partnership: CGIAR and NARES Leading Dryland Innovation for Impact
- - Pathways to power in fragile settings: rethinking women’s roles in agriculture and food systems
- - Integrated transcriptomics and metabolomics analyses provide new insights into cassava in response to nitrogen deficiency
- - Study Reveals How Bacteria Bypass Plant Defenses
- - Long-term Study Shows GHG Emissions from Agricultural Soils
- - The long-term straw return resulted in significant differences in soil microbial community composition and community assembly processes between wheat and rice
- - Study Reveals Narrow Genetic Basis of European Potato
- - Pangenome Reveals Genetic Diversity, Evolution, and Domestication of Rice
- - Bacterial pathogen deploys the iminosugar glycosyrin to manipulate plant glycobiology
- - Late Blight Field Resistance in Potatoes With Genes from Wild Relative
- - Experts Recommend Policy Strategies to Support Bt Cotton Adoption in India
- - Late blight field resistance in potatoes carrying Solanum americanum resistance genes (Rpi-amr3 and Rpi-amr1)
- - CABBI Team Accelerates Plant Bioengineering Using Robotics Lab
- - Researchers Develop Transgene-free Gene-edited Poplar Trees
- - A comprehensive all-in-one CRISPR toolbox for large-scale screens in plants
- - Gen Zs Lead Strong Support for Gene Editing in UK
- - Nanotechnology Plays Vital Role in Gene Editing
- - Association mapping of haploid male fertility in sweet corn
- - Experts Develop All-in-one CRISPR Toolbox for Large-scale Screens in Plants
- - Gene Editing Improves Fruit Quality of Tomatoes
- - Water stress enhances triacylglycerol accumulation via different mechanisms in wild-type and transgenic high-leaf oil tobacco
- - Water Stress Improves Triacylglycerol Production in High-leaf Oil Tobacco Plants
- - Genome Sequences for Five Duckwood Species Reveal Potential for Food and Fuel Source
- - Duckweed genomes and epigenomes underlie triploid hybridization and clonal reproduction
- - FSANZ Approves First Cell-Cultured Food to be Marketed and Sold in Australia and New Zealand
- - Research Reveals Cry1Ab Kills Pests Through Two Pathways; Increasing Sustainability of GE Crops
- - Two genetically modified insect-resistant maize events reduced fumonisinpollution under the stress of Lepidoptera in China
- - ISAAA Webinar Explores Animal Biotech Regulations in the Philippines
- - Scientists Develop New Gene Editing Tool that Replaces Entire Genes
- - Chromosomal structural variation loci HSS1 and HSS6 lead to hybrid sterility in rice
- - Lettuce Biofortified With Folate Stable in Field Conditions
- - Two Insect-resistant Maize Events Reduce Fumonisin Pollution in China
- - Genome-wide comparison reveals large structural variants in cassava landraces
- - At FAO Council, Director-General QU Dongyu emphasizes world`s growing need for the urgent and accelerated transformation of global agrifood systems
- - Earthquake in Myanmar: As the planting season nears, FAO scales up emergency response for farmers
- - Temporal transcriptome analysis reveals the two-phase action of florigens in rice flowering
- - FAO - 177th Session of the FAO Council Closing Remarks
- - MEDIA RELEASE | New CGIAR flagship report translates insights into impact for food systems in crisis
- - A Golden2-like transcription factor regulates Brassica napus seed vigor after artificial aging
- - OPINION | Future of our agriculture lies in science
- - Harvesting Collaboration for Food Security. CGIAR Science Week Closes.
- - Crop pest responses to global changes in climate and land management
- - Experts Review Crop Pests` Responses to Climate and Land Management Changes
- - Engineered Yeast Converts Methanol to D-lactic Acid
- - A major QTL region associated with powdery mildew resistance in leaves and fruits of the reconstructed garden strawberry
- - New Technique to Unlock Sorghum`s Huge Climate Change Potential
- - Improved Maize Varieties Boost Yields and Farm Income
- - Identification and analysis of miRNA - mRNA regulatory modules associated with resistance to bacterial leaf streak in rice
- - First Skin-Transplant Potato Gets Plant Breeder`s Rights in The Netherlands
- - Experts Develop COVID-19 Vaccine from Rice
- - Rice-derived SARS-CoV-2 glycoprotein S1 subunit vaccine elicits humoral and cellular immune responses
- - Treating NGTs as Conventional Products May Improve Market Acceptance
- - Switzerland Drafts New Law on NBTs
- - A conserved ARF–DNA interface underlies auxin-triggered transcriptional response
- - Study Shows US Public Attitudes on Genetically Engineered Microbiomes
- - Pistachio DNA Map Could Lead to More Nutritious and Sustainable Nuts
- - Natural vs. genetically engineered microbiomes: understanding public attitudes for indoor applications and pathways for future engagement
- - Australia`s Gene Technology Regulator Invites Comments on GM Canola Field Trial
- - FSA and FSS Confirm Safety of GM Soybean MON 87705 x MON 87708 x MON 89788
- - CRISPR-enhanced iron accumulation in TBR225 rice via OsNRAMP7 overexpression
- - Engineered Red Yeast Converts Forestry Waste into Valuable Fatty Acid
- - Cell-Cultivated Fish Diminishes Seafood Allergy Risks
- - Improving yield-related traits by editing the promoter and distal regulatory region of heading date genes Ghd7 and PRR37 in elite rice variety Mei Xiang Zhan 2
- - Iron-biofortified Rice Developed to Help Combat Iron Deficiency Anemia
- - Unlocking the Potential of Animal Biotech in the Philippines: Research Updates and Regulatory Prospects (10/4/2025)
- - A genome-wide association study prioritizes VRN1-2 as a candidate gene associated with plant height in soybean
- - ICRISAT Successfully Conducts National Intellectual Property Yatra in Assam
- - ICRISAT Celebrates its 54th Foundation Day with a Vision for Resilient and Prosperous Drylands
- - Mechanism of microplastics in the reduction of cadmium toxicity in tomato
- - GIZ and IRRI partnership demonstrates a future of sustainable agriculture in Vietnam
- - Reducing ambiguity in agricultural research and development: Insights from a global experiment with plant breeding professionals
- - Multi-Omics Analysis Reveals That AhNHL Contributes to Melatonin-Mediated Cadmium Tolerance in Peanut Plants
- - IFAD at the 2025 N4G Paris Summit
- - International Rice Research Institute (IRRI) and the Department of Agricultural Extension (DAE) strengthen their partnership to enhance rice research and innovation in Bangladesh
- - Genome-wide analysis of KCS genes in tomato and functional characterization of SlKCS8 and SlKCS10 in drought tolerance
- - Participatory foresight analysis to identify near-market segments for cowpea in Western Africa
- - Market intelligence area of work expands reach with four communication tools
- - Cas12a-knock-in mice for multiplexed genome editing, disease modelling and immune-cell engineering
- - The genetic diversity of our plants and forests is at risk, new FAO reports warn
- - Conflict and rising food prices drive Congolese into one of the world`s worst food crises according to new IPC data
- - Auxin-responsive OsMADS60 negatively mediates rice tillering and grain yield by modulating OsPIN5b expression
- - Nature`s groceries and safety net: how forests help feed the world better
- - Yale University Introduces Cas12a-Based Method for Dual Gene Activation and Knockout
- - TaFT-D1 positively regulates grain weight by acting as a coactivator of TaFDL2 in wheat
- - OsMADS60 Controls Rice Yield Through Auxin Transport
- - Researchers Identify Gene Regulating Grain Weight in Wheat
- - Melatonin enhances salt tolerance by promoting CcCAD10-mediated lignin biosynthesis in pigeon pea
- - CRISPR-Cas9 Helps Enhance Cassava`s Disease Resistance, Drought Tolerance, and Starch Biosynthesis
- - University of Ottawa Students Grow Anti-diabetic Drugs in Plants
- - OsbZIP27 coordinates with OsHUB1 and OsHUB2 to modulate drought tolerance in rice
- - Study Confirms Safety of GM Soybean in a Subchronic Rodent Feeding Study
- - EFSA GMO Panel Releases Scientific Opinion on GM Mazie DAS1131
- - Exogenous Melatonin Improves Drought Tolerance by Regulating the Antioxidant Defense System and Photosynthetic Efficiency in Fodder Soybean Seedings
- - UC Davis Develops New Genetic Tool for Shorter Wheat
- - Australia`s Gene Technology Regulator Invites Comments on Field Trial of GM Safflower
- - Food safety assessment of genetically modified soybean DBN9004×DBN8002×DBN8205 in a subchronic rodent feeding study
- - Mexico Issues Constitutional Decree Banning GM Corn
- - South Korea Confirms Cultivation Safety of GM Potatoes
- - Agriculture`s impact on water–energy balance varies across climates
- - Creating a Network of Aflatoxin Management Champions in the Global South
- - ICRISAT Successfully Conducts National Intellectual Property Yatra in Assam
- - Genome-wide association study reveals genetic loci for seed density per silique in rapeseed (Brassica napus L.)
- - NL-CGIAR info-session on the Scaling for Impact Program organized by the Netherlands Food Partnership (NFP)
- - CGIAR and ICRISAT Drive Gender Equality in South-South Training Program
- - Maize B chromosome affects the flowering time
- - Preparing for COP30: Asia Pacific drives action towards climate targets
- - Fertilize Right combines science and partnership to transform Vietnam`s rice production
- - The SUbventral-Gland Regulator (SUGR-1) of nematode virulence
- - International Day of Forests 2025: Engaging the next generation in the connection between forests and foods
- - FSA Review Explores Consumer Responses to Cell-Cultivated Products
- - Evaluating Xanthomonas oryzae pv. oryzae (Xoo) infection dynamics in rice for distribution routes and environmental reservoirs by molecular approaches
- - Report Projects Boost in Demand from GM Food Market in Upcoming Years
- - Scientists Sequence Wild Barley Genome; Paving the Way for Climate-Resilient Crops
- - Identification of candidate genes associated with resistance to aflatoxin production in peanut through genetic mapping and transcriptome analysis
- - Researchers in Japan Develop Genome Editing Method that Partially Inihibits Gene Function
- - Experts Discover New CRISPR-Cas Systems
- - Sequential evolution of resistance by western corn rootworm to multiple Bacillus thuringiensis traits in transgenic maize
- - Australia`s Gene Technology Regulator Approves Field Trial of GM Sorghum
- - EU Council Agrees Negotiating Mandate for NGTs
- - Gene-Lifestyle Interactions in Renal Dysfunction: Polygenic Risk Modulation via Plant-Based Diets, Coffee Intake, and Bioactive Compound Interactions
- - Adverse climatic conditions drive coffee prices to highest level in years
- - Harlem Globetrotters named FAO Global Goodwill Ambassadors
- - Precise genome editing of Dense and Erect Panicle 1 promotes rice sheath blight resistance and yield production in japonica rice
- - Understanding cowpea anthracnose in Nigeria: New findings on its causal agents
- - Gene Editing Leads to Rice Sheath Blight Resistance and Yield Increase
- - RNAi and genome editing of sugarcane: Progress and prospects
- - Study Reveals Gene that Reduces Wheat Grain Weight
- - Experts Review Applications of TALENs in Sugarcane
- - Genetic dissection of flowering time and fine mapping of qFT.A02-1 in rapeseed (Brassica napus L.)
- - Scientists Develop Gene-Edited Lettuce to Fight Micronutrient Deficiencies
- - Nigeria Becomes the 80th Member of the International Union for the Protection of New Varieties of Plants
- - Papain expression in the Escherichia coli cytoplasm by T7-promoter engineering and co-expression with human protein disulfide isomerase (PDI) and thiol peroxidase (GPx7) genes
- - Scientists Have Identified Genetic Defense Against Soybean Cyst Nematodes
- - Researchers Identify Genes that Help Sorghum Resist Striga by Preventing its Germination
- - Exploring resistance mechanisms and identifying QTLs for brown planthopper in tropical and subtropical rice (Oryza sativa L.) germplasm
- - Report Tackles China`s Agricultural Transformation Plans for 2025
- - Brazilian Fish and CAT Introduce First Gene-Edited Tilapia in Brazil
- - Resistance to Striga parasitism through reduction of strigolactone exudation
- - Genetic Insights Reveal How Plants Adapt to Climate Change
- - ISAAA and DA Biotech Program to Hold a Webinar on Regulatory Frameworks for New Breeding Techniques in Crops
- - The evolutionary history of the common bean (Phaseolus vulgaris) revealed by chloroplast and nuclear genomes analysis
- - What does process improvement look like for CGIAR crop breeding programs?
- - Drylands under pressure: Science and solutions for global stability
- - tRNA selectivity during ribosome-associated quality control regulates the critical sterility-inducing temperature in two-line hybrid rice
- - Director-General QU Dongyu - Bilateral meeting with the Prime Minister of Ethiopia, H.E. Abiy Ahmed
- - Scientists Review RNAi-induced Effects in GM Plants
- - CAS9 Mediated In-Planta Defence Strategy Against Tomato Leaf Curl New Delhi Virus (ToLCNDV) in Tomato
- - Fewer Off-Target Gene Editors Using AI
- - Scientists Use Gene Editing to Combat Tomato Leaf Curl New Delhi Virus
- - Genome-wide association study to identify candidate genes for submergence tolerance during rice seed germination
- - CRISPR Sheds Light on Enhancing Nitrogen Fixation in Bean Genes
- - Scientists Develop Gene Editing Method to Reduce Corn Plant Height
- - Magnaporthe oryzae infection triggers rice resistance to brown planthopper through the influence of jasmonic acid on the flavonoid biosynthesis pathway
- - IRRI and smart agriculture technology leader XAG collaborate to promote digital agriculture and precision farming in the Philippines
- - Japan contributes $11.9 million to scale up FAO`s emergency and resilience activities
- - Leaky ribosomal scanning enables tunable translation of bicistronic ORFs in green algae
- - Somalia: Drought, conflict and high food prices risk pushing 4.4 million people into hunger, the Federal Government of Somalia and UN warn
- - INTERNATIONAL WOMAN DAY IWD 2025 campaign theme is `Accelerate Action`
- - Integrating phenomic selection using single-kernel near-infrared spectroscopy and genomic selection for corn breeding improvement
- - Nature-Positive Solutions: Transforming Agrifood Systems for Resilience and Sustainability
- - Empowering women in livestock: addressing gender barriers in Uganda`s pig sector
- - Mapping and functional characterization of the golden fruit 1 (gf1) in melon (Cucumis melo L.)
- - With Science, We Can: Degraded Lands Transform into Productive Farms in Latur, India
- - CBD COP16.2 Side Event: “Meeting people`s needs through sustainable use of biodiversity within agrifood systems
- - Knocking out artificially selected gene GmAOC4H8 improves germination in soybean
- - Study reveals Africa will reach 1.5°C climate change threshold by 2040 even under low emission scenarios
- - Experts Pinpoint Rice Proteins Vital in Drought Stress Tolerance and Yield
- - Design of CoQ10 crops based on evolutionary history
- - Chinese Scientists Use Gene Editing to Develop CoQ10-Producing Rice
- - Genome-Edited Rice Shows Resistance to Bacterial Blight in East Africa
- - Creation of fragrant peanut using CRISPR/Cas9
- - Scientists Use CRISPR to Develop Fragrant Peanuts
- - Genetic Engineering Improves Salt Tolerance in Wheat
- - Development and Application of Pik Locus-Specific Molecular Markers for Blast Resistance Genes in Yunnan Japonica Rice Cultivars
- - Rubber Growers Call for GM Rubber Approval to Address Price Slump and Pest Attacks
- - Australian OGTR Receives License Application for Commercial Release of GM Cotton
- - A wild-allele GsPP2C-51-a1 enhances tolerance to drought stress in soybean and Arabidopsis
- - Study Explores Impact of Bt Cottonseed Cake on Growth and Health of Rams
- - Scientists Decode the Genome of White Oak
- - Brems1 mutation induced tapetum deficiency leading to male sterility in Chinese cabbage (Brassica rapa L. ssp. pekinensis)
- - Researchers Find Corn Lines with High Flavonoid Content Could Kill Major Crop Pest
- - Study Reveals Consumer Acceptance of Gene-Edited Food
- - Strategies and Protocols for Optimized Genome Editing in Potato
- - NARES and IRRI advance market-driven varieties and accelerate adoption of new rice varieties in Africa
- - IRRI receives Cross of Friendship medal from Lao PDR
- - First Report of Lasiodiplodia theobromae Causing Wilt and Fruit Rot of Pepper in Hainan Province, China
- - G20: Food security is vital for peace, stability and human dignity, FAO says
- - UN Biodiversity Conference COP16 talks resume in Rome: What`s at stake?
- - DArTseq-based silicoDArT and SNP markers reveal the genetic diversity and population structure of Kenyan cashew (Anacardium occidentale L.) landraces
- - NATURE+ in India: Country Report 2022-2024
- - NATURE+ in Vietnam: Country report 2022-2024
- - Ethylene response factor SlERF.D6 promotes ripening in part through transcription factors SlDEAR2 and SlTCP12
- - Engineered Animals to Fight Mercury Pollution
- - Study Elucidates Salt Tolerance Breeding in Plants
- - Papain expression in the Escherichia coli cytoplasm by T7-promoter engineering and co-expression with human protein disulfide isomerase (PDI) and thiol peroxidase (GPx7) genes
- - Scientists Have Identified Genetic Defense Against Soybean Cyst Nematodes
- - Researchers Identify Genes that Help Sorghum Resist Striga by Preventing its Germination
- - Exploring resistance mechanisms and identifying QTLs for brown planthopper in tropical and subtropical rice (Oryza sativa L.) germplasm
- - Report Tackles China`s Agricultural Transformation Plans for 2025
- - Brazilian Fish and CAT Introduce First Gene-Edited Tilapia in Brazil
- - Resistance to Striga parasitism through reduction of strigolactone exudation
- - Genetic Insights Reveal How Plants Adapt to Climate Change
- - ISAAA and DA Biotech Program to Hold a Webinar on Regulatory Frameworks for New Breeding Techniques in Crops
- - The evolutionary history of the common bean (Phaseolus vulgaris) revealed by chloroplast and nuclear genomes analysis
- - What does process improvement look like for CGIAR crop breeding programs?
- - Drylands under pressure: Science and solutions for global stability
- - tRNA selectivity during ribosome-associated quality control regulates the critical sterility-inducing temperature in two-line hybrid rice
- - Director-General QU Dongyu - Bilateral meeting with the Prime Minister of Ethiopia, H.E. Abiy Ahmed
- - Scientists Review RNAi-induced Effects in GM Plants
- - CAS9 Mediated In-Planta Defence Strategy Against Tomato Leaf Curl New Delhi Virus (ToLCNDV) in Tomato
- - Fewer Off-Target Gene Editors Using AI
- - Scientists Use Gene Editing to Combat Tomato Leaf Curl New Delhi Virus
- - Genome-wide association study to identify candidate genes for submergence tolerance during rice seed germination
- - CRISPR Sheds Light on Enhancing Nitrogen Fixation in Bean Genes
- - Scientists Develop Gene Editing Method to Reduce Corn Plant Height
- - SEGS-1 episomes generated during cassava mosaic disease enhance disease severity
- - GM Maize MON810 Shows No Adverse Effects on Surface-active Springtails
- - Key Wheat Genes Confer Resistance Against Powdery Mildew
- - Evolutionary signatures of the erosion of sexual reproduction genes in domesticated cassava (Manihot esculenta)
- - Global Survey Reveals Public Sentiment Towards Transformative Technologies in Health and Agriculture
- - University of Nevada, Reno Researcher Develops Sorghum Varieties for Dairy Feed and Gluten-Free Food
- - Effects of genetically modified maize MON810 on surface-active springtails (Collembola)
- - Mexico Cancels GM Corn Restrictions, Follows USMCA Ruling
- - Field Trial of Gene-Edited Wheat in UK Gets ACRE Approval
- - Combining Controlled-Release and Normal Urea Enhances Rice Grain Quality and Starch Properties by Improving Carbohydrate Supply and Grain Filling
- - The disappearing savannas: Is West Africa trading soil health for farmland?
- - How do food and fertilizer price spikes and volatility impact Central America and the Caribbean?
- - Generation of novel bpm6 and dmr6 mutants with broad-spectrum resistance using a modified CRISPR/Cas9 system in Brassica oleracea
- - Experts Develop Engineered Yeast with Increased Healthy Fatty Acid
- - ZQTALEN: A Simple and Efficient Gene Editing Tool for Plants
- - The candidate gene SibHLHA regulates anthocyanin-driven purple pigmentation in Sesamum indicum flowers
- - Sustainable Agriculture Contributes to Achieving Zero Hunger
- - Paper Examines Global Crop Export Trade of GM Technology
- - The TCP transcription factor TAC8 positively regulates the tiller angle in rice (Oryza sativa L.)
- - USDA APHIS Reinstates Notifications for Biotech Products
- - Gene from Agrobacterium Confers Drought Tolerance in Wheat
- - Advancing sustainable agriculture for goal 2: zero hunger - a comprehensive overview of practices, policies, and technologies
- - ISAAA Webinar Tackles Novel Foods for Sustainable Agriculture
- - Poland Seeks to Break EU Deadlock on NGT Proposal
- - BrCYP71 mutation resulted in stay-green in pak choi (Brassica rapa L. ssp. chinensis)
- - CABBI Researchers Develop Oil-Rich Sorghum
- - LBC and ISAAA to Hold 8th International Livestock Biotechnology Symposium
- - Adaptive evolution and mechanism elucidation for ethanol tolerant Saccharomyces cerevisiae used in starch based biorefinery
- - Nitrogen use efficiency must be improved to reduce harm to human and environmental health
- - Food insecurity deepens in Lebanon following conflict, new report shows
- - Genome-wide association studies unveils the genetic basis of cell wall composition and saccharification of cassava pulp
- - FAO celebrates Chinese Lunar New Year with focus on transformation and call for global solidarity
- - Design workshop lays foundation for new training program on food systems governance for sustainable healthy diets in Bangladesh
- - Overexpression of the persimmon ABA receptor DkPYL3 gene alters fruit development and ripening in transgenic tomato
- - How conflict drives hunger: Six channels through the food system
- - NATURE+ supports Kenyan smallholders with equipment, training for value-added agrobiodiversity production
- - Mycotoxin concentrations in rice are affected by chalkiness, grain shape, processing type, and grain origin
- - Researchers Test Dual-process Framework for Understanding Acceptance of Genetic Modification
- - ARS Research Team Boost Alfalfa`s Salt Tolerance
- - Engineering source-sink relations by prime editing confers heat-stress resilience in tomato and rice
- - Study Explores Need for Confined Field Trials for Rapeseed in Japan
- - Defra Secretary Confirms Timetable for Precision Breeding Act
- - Comparative genomic prediction of resistance to Fusarium wilt (Fusarium oxysporum f. sp. niveum race 2) in watermelon: parametric and nonparametric approaches
- - UMass Amherst Team to Develop New Plant-based Protein Food from Chickpeas and Peas
- - Gene-edited Soil Bacteria Could Provide More Nitrogen for Corn
- - Do confined field trials add value for the environment risk assessment of genetically modified Brassica napus L. in Japan?
- - USDA ERS Report Shows Recent Trends on GE Crop Adoption in the US
- - Plate of the Future: Nutritious and Sustainable Novel Foods
- - Transcriptomic analysis reveals pathogenicity mechanisms of Phytophthora capsici in black pepper
- - IRRI and smart agriculture technology leader XAG collaborate to promote digital agriculture and precision farming in the Philippines
- - Partnerships and co-investment drive sustainable impact of Chan-henh project in Northwest Highlands of Vietnam
- - Investigation of the physicochemical factors affecting the in vitro digestion and glycemic indices of indigenous indica rice cultivars
- - Study Pinpoints Regulator of Cold Stress Tolerance in Rice
- - CAAS Develop Gene Editing System for Wheat
- - OsNCED5 confers cold stress tolerance through regulating ROS homeostasis in rice
- - Researchers Identify Gene Controlling Growth and Development in Bananas
- - Experts Use Genome Editing to Make Tomatoes Yield Earlier
- - MaGA20ox2f, an OsSD1 homolog, regulates flowering time and fruit yield in banana
- - Genomes of Alopecurus Grasses Shed Light on Weed Resistance
- - Survey Shows Public Sentiment on Microbiome Engineering
- - The interaction of Serratia bacteria and harmonine in harlequin ladybird confers an interspecies competitive edge
- - Gene-silencing Spray to Fight Fusarium Head Blight in Cereal Crops
- - PhilRice Study Pinpoints Low-GI Rice Varieties for Diabetics
- - Intra species dissection of phytophthora capsici resistance in black pepper
- - CSIRO and Oxitec to Target Invasive Pests Across Australia and Oceania
- - Bt to Help Combat Devastating Citrus Greening Disease
- - Tests for segregation distortion in tetraploid F1 populations
- - Expected Profitability and Perception Drive Farmer`s Participation in GM Crop Farming
- - 153 Nobel and World Food Prize Laureates Issue Urgent Wake-Up Call Over Hunger Tipping Point
- - The IAS Research Activities In 2024
- - Microsatellite markers development and molecular fingerprinting of cashew cultivars
- - A jack of all fruits
- - New report highlights critical food system trends and challenges in countdown to 2030
- - Genome-wide profile analysis of the Hsp20 family in lettuce and identification of its response to drought stress
- - FAO Director-General visits China, reaffirming the importance of partnerships on Science and Innovation
- - Empowering Ethiopian livestock keepers: A new approach to learning
- - Development and characterization of a complete set of monosomic alien addition lines from Raphanus sativus in Brassica oleracea
- - Connecting the Dots: Linking Social Protection to Climate Change
- - Advancing Odisha`s Millet Mission: A Science-Policy Workshop Highlighting Transformative Pathways
- - NF-YC15 transcription factor activates ethylene biosynthesis and improves cassava disease resistance
- - Accelerating Agricultural R&D Transfer Enhances Global Food Security
- - FSANZ Calls for Comment on Food from New GM Soybean
- - Carbon gain in upper but loss in deeper cropland soils across China over the last four decades
- - Study Concludes GM Maize DP-915635-4 As Safe As Conventional Counterpart
- - Australian OGTR Invites Public Comments for Field Trial of GM Sorghum
- - GWAS analysis revealed genomic loci and candidate genes associated with the 100-seed weight in high-latitude-adapted soybean germplasm
- - ISAAA GM Approval Database Feedback Survey
- - GM Mosquitoes with Toxic Semen Offer Faster Solution to Control Mosquito Populations
- - Agrobacterium-Mediated Transformation and Targeted Mutagenesis Using SpCas12f in Rice
- - Study on GM Rice to Cure Hay Fever Moves Forward
- - China Issues New and Renewed Biosafety Certificates for GE and Gene-edited Crops
- - Vietnam Agriculture In 2024: Accelerate Creativity & Sustainable Efficiency
- - Genome Resequencing for Autotetraploid Rice and Its Closest Relatives Reveals Abundant Variation and High Potential in Rice Breeding
- - Inclusive and equitable canal water management towards sustainable agriculture urged at Bangladesh policy dialogue
- - Hybrid rice + innovations: a path forward for climate-smart agriculture
- - Dissecting the cellular architecture and genetic circuitry of the soybean seed
- - How a Senegal fishing family found safe harbour at home
- - Climate-smart irrigation reaps rewards for rural women
- - PsDMAP1/PsTIP60-regulated H4K16ac is required for ROS-dependent virulence adaptation of Phytophthora sojae on host plants
- - FAO calls for urgent action to address widening famine in Sudan
- - Training civil servants to tackle climate-induced migration challenges in Guatemala
- - Fine mapping of the Chilli veinal mottle virus resistance 4 (cvr4) gene in pepper (Capsicum annuum L.)
- - Towards a Climate Security Observatory 2.0: lessons from a year of implementation
- - Promoting mechanized farming technologies in Mbire and Murewa through the Agroecology Fairs
- - Expression pattern of Stlhcb gene family in potato and effects of overexpression of Stcp24 gene on potato photosynthesis
- - Business-to-business workshops promote technology uptake among agripreneurs in Tanzania
- - Addressing climate risks for Nigerian pastoralists
- - Transcriptomic study of the role of MeFtsZ2-1 in pigment accumulation in cassava leaves
- - Milestone: COVID-19 five years ago
- - Transitioning Farming Systems for Resilience and Food Security in Malawi
- - Unlocking ABA`s role in rice cold tolerance: insights from Zhonghua 11 and Kasalath
- - FAO publishes new guidelines for surveillance of influenza in cattle
- - FAO Food Price Index dips during the month of December
- - Heterosis for Resistance to Insect Herbivores in a 3-Line Hybrid Rice System
- - From space to soil: Advancing crop mapping and ecosystem insights for smallholder agriculture in Kenya
- - Elite domination of freshwater canals in coastal Bangladesh adversely affects local communities and livelihoods
- - A frameshift mutation in JAZ10 resolves the growth versus defense dilemma in rice
- - Future market segment: Can sweet sorghum power the next generation of clean energy?
- - What do we know about the future of agrifood systems in South Asia?
- - OsNAC3 regulates seed germination involving abscisic acid pathway and cell elongation in rice
- - Opportunities for the seed sector to contribute to nutrition, sustainability, and livelihoods tackled at NSC 2024
- - Technical Platform on the Measurement and Reduction of Food Loss and Waste
- - A GWAS identified loci and candidate genes associated with fiber quality traits in a new cotton MAGIC population
- - How does social assistance affect consumption and diets after flooding in Southern Bangladesh?
- - The wonder and importance of biological control in farming and agrifood systems
- - A key ABA biosynthetic gene OsNCED3 is a positive regulator in resistance to Nilaparvata lugens in Oryza sativa
- - Opportunities for the seed sector to contribute to nutrition, sustainability, and livelihoods tackled at NSC 2024
- - Food and nutrition crisis deepens across Sudan as famine identified in additional areas
- - Toward systems agroecology: Design and control of intercropping
- - Farmer-centred approaches for turning biodiversity commitments into actions
- - Revisiting the 2006 Abuja Fertilizer Declaration with Nitrogen use efficiency and yield-gap lenses
- - OsWRKY49 on qAT5 positively regulates alkalinity tolerance at the germination stage in Oryza sativa L. ssp. japonica
- - Takaful program: Giving Egypt`s poor a hand up, not just a handout
- - Integrating nutrition-sensitive agricultural systems into national policy frameworks: Lessons from Vietnam
- - miR396b/GRF6 module contributes to salt tolerance in rice
- - Driving Farmer-Centered Digital Transformation in African Agriculture ICRISAT and KALRO Lead Stakeholder Workshop on Advancing Digital Agriculture in Nairobi
- - Enhancing access to quality seed for farmers in Burundi
- - The analysis of the genetic loci affecting phenotypic plasticity of soybean isoflavone content by dQTG.seq model
- - Fighting nematodes with banana paper: An approach to boost potato yields in Kenya
- - Empowering Community Rangeland Health Workers: A Novel Path to Sustainable Ecosystem Management in Ethiopia
- - The OsNAC41-RoLe1-OsAGAP module promotes root development and drought resistance in upland rice
- - Advancement in Rural Digital Infrastructure: Charting the Way Forward for Digital Agriculture
- - Low GI rice seen as promising solution to mitigate Asia`s diabetes crisis
- - Mutations in a Leucine-Rich Repeat Receptor-Like Kinase gene result in male sterility and reduction in the number and size of fruit warts in cucumber (Cucumis sativus L.)
- - Global Environment Facility approves $68 million for agrifood systems solutions
- - Director-General urges heads of FAO country offices to take collective action to the next level
- - Origin and evolution of auxin-mediated acid growth
- - Unlocking the potential of underutilized legumes: IITA-CGIAR and SUL lead strategic conference for sustainable agricultural innovation
- - Agricultural Emergency Response with Potato and Sweetpotato Technologies for Livelihood Restoration of Communities Affected by Conflict
- - Calcium signaling triggers early high humidity responses in Arabidopsis thaliana
- - EFSA Releases Scientific Opinion on the Food Enzyme Endo-1,4-β-xylanase from GM Bacillus subtilis strain AR-153
- - Scientists Develop Breeding Strategies to Create Climate-smart Crops
- - Genome-wide association study of haploid female fertility (HFF) and haploid male fertility (HMF) in BS39-derived doubled haploid maize lines
- - GM Soybeans Show No Adverse Effects on Rat Health and Gut Microbiota
- - Australian OGTR Receives License Application for Field Trial of GM Canola
- - Map-based cloning revealed BhAPRR2 gene regulating the black peel formation of mature fruit in wax gourd (Benincasa hispida)
- - New Zealand Releases Proposed Updates on Gene Technology Framework
- - IGI Leads Development of Technical Approach to Global Plant Genome Editing Regulation
- - Gene Profiling in Late Blight Resistance in Potato Genotype SD20
- - Global forest products facts and figures 2023 shows fall in global trade in wood and paper products
- - FAO launches first major global assessment of salt-affected soils in 50 years
- - Advancements and Prospects of Genome-Wide Association Studies (GWAS) in Maize
- - Director-General urges heads of FAO country offices to take collective action to the next level
- - Peskas: The fisheries monitoring system making waves in Asia and Africa
- - Soybean steroids improve crop abiotic stress tolerance and increase yield
- - IRRI eyes stronger, long-term sustainable rice and climate action partnership with Thailand
- - New innovation integrates water resources management in Cambodia`s Tonle Sap floodplain and Mekong delta
- - The heat shock factor 20-HSF4-cellulose synthase A2 module regulates heat stress tolerance in maize
- - Gene Editing Market Value to Reach USD 17.5 B by 2031
- - CRISPR-Cas9 for Disease Resistant Vines
- - Reduced content of gamma-aminobutyric acid enhances resistance to bacterial wilt disease in tomato
- - Gene Editing and Plant Domestication as Key Tools for Food Security and Facing Climate Change
- - CRISPR Enhances Resistance of Tomato Against Bacterial Wilt Disease
- - Breeding for brown plant hopper resistance in rice: recent updates and future perspectives
- - Research Reveals Warming Nighttime Temperatures Reduce Rice Quality in East Asia
- - Japan Remains a Key GM Product Import Market
- - Pyramiding BPH genes in rice maintains resistance against the brown planthopper under climate change
- - US Court Overturns USDA Ruling on Genetically Engineered Plants
- - DA BPO Names the Awardees of the 9th Filipino Faces of Biotechnology
- - Electrocatalytic nitrate reduction using iron single atoms for sustainable ammonium supplies to increase rice yield
- - "Booster" Gene Game-Changer for Plant-Based Jet Fuel and Food Production
- - ISAAA Webinar Explores the Science and Application of Gene Drives
- - Genome-wide association study and genomic prediction for resistance to brown planthopper in rice
- - Experts Develop Heatwave-tolerant Potatoes
- - Kenya`s National Biosafety Authority Opens Public Comments on the Proposed Environmental and Market Release of Bt Maize
- - Chronologically inappropriate morphogenesis (Chinmo) is required for maintenance of larval stages of fall armyworm
- - GM Crop Market Projected to Reach USD 36 Billion by 2031
- - New Study Investigates Impact of Marketing Expectations on Golden Rice Purchase Intentions in Bangladesh and the Philippines
- - Novel SNP markers and other stress-related genomic regions associated with nitrogen use efficiency in cassava
- - International Research Team Unravels Wheat`s Genetic Past to Transform its Future
- - What do you think of this newsletter?
- - Yield determination of temperate maize hybrids with different end-uses: An ecophysiological analysis
- - Scientists Reveal Hidden DNA in Plants Play Crucial Role in Photosynthesis
- - Global Crop Yields Have Grown Steadily in the Last Six Decades
- - A novel Pik allele confers extended resistance to rice blast
- - Director-General QU Dongyu: 176th Session of the FAO Council Opening Statement (2/12/2024)
- - ICRISAT Co-Releases First Pearl Millet and Sorghum Hybrids in Zimbabwe Science-led innovation is reshaping agriculture in Zimbabwe, directly addressing critical challenges such as climate change, food insecurity, and nutritional deficits.
- - CRISPR–Cas9-mediated promoter editing of FERONIA-Like receptor 13 increases plant growth and disease resistance in rice
- - Collaboration and Learning in Action: A Look into Colombia`s Food Systems Community of Practice
- - Promoter Editing of FLR13 Boosts Growth and Disease Resistance in Rice
- - Development of genetically modified rust resistant wheat: A breakthrough by dinted introgression of novel DREB2C and HSFA2 genes under stress induced expression
- - Heat Shock Protein Helps Maize Plants Resist Drought
- - genXtraits to Use Fulcrum™ Toolkit from Pairwise to Develop High-Value Enhanced Crops
- - ZmDnaJ-ZmNCED6 module positively regulates drought tolerance via modulating stomatal closure in maize
- - Researchers Develop Rust Resistant Wheat with Increased Heat Tolerance
- - Experts Develop GM Live Vaccine to Protect Cattle from Anaplasma Marginale
- - Analytical prediction of genetic contribution across multiple recurrent backcrossing generations
- - Biologists Show Two Genes Work Together to Trigger Rice Embryo Formation
- - Chickpea Heat Tolerance Traits Identified by Australian Researchers
- - Cashew nut (Anacardium occidentale L.) and cashew nut oil reduce cardiovascular risk factors in adults on weight-loss treatment: a randomized controlled three-arm trial (Brazilian Nuts Study)
- - Researchers Identify Genetic Secrets of High-Yield Fuji Apple
- - New Gene Drive Technology Reverses Insecticide Resistance in Pests
- - QTL-seq and QTL mapping identify a new locus for Cercospora leaf spot (Cercospora canescens) resistance in mungbean (Vigna radiata) and a cluster of Receptor-like protein 12 (RLP12) genes as candidate genes for the resistance
- - ISAAA Inc. Joins National Biotechnology Week in the Philippines
- - Leading Breakthroughs: Gene Drives for a Sustainable Agriculture and Biodiversity Conservation
- - KnockTF 2.0: a comprehensive gene expr profile database with knockdown / knockout of transcription (co-)factors in multiple species
- - ICRISAT Open Field Day Showcases High-Yield Seeds and Climate-Resilient Innovations in Mali
- - Scaling sustainable rice farming and nutrition-focused resilience in Asia-Pacific: A COP29 Call to action
- - Genetic variation in a heat shock transcription factor modulates cold tolerance in maize
- - IRRI`s Methane Accelerator for Southeast Asia (MASEA) Takes Off
- - Democratic Republic of the Congo: FAO urges greater focus on humanitarian needs of vulnerable populations and implementation of at-scale programming to strengthen resilience against recurrent shocks
- - The RING-type E3 ligase RIE1 sustains leaf longevity by specifically targeting AtACS7 to fine-tune ethylene production in Arabidopsis
- - FAO-WHO Codex Alimentarius Commission adopts new standards
- - FAO urges all its Members and partners to join the Global Alliance against Hunger and Poverty
- - Genome-wide identification and expression analyses of CYP450 genes in sweet potato (Ipomoea batatas L.)
- - General Statement of CGIAR participation at COP29 in Baku, Azerbaijan
- - COP29 Key Take Aways, Negotiations, Side Events and Coverage
- - Releasing a sugar brake generates sweeter tomato without yield penalty
- - Report Highlights Gene Editing Regulations in Australia
- - EFSA GMO Panel Releases Scientific Opinion on Soy Leghemoglobin Produced from GM Komagataella phaffii
- - Genomic resources, opportunities, and prospects for accelerated improvement of millets
- - Scientists Produce Sweeter Tomatoes Without Yield Penalty
- - International Research Team Publishes Barley Pangenome
- - Fine mapping and functional validation of the candidate gene BhGA2ox3 for fruit pedicel length in wax gourd (Benincasa hispida)
- - Scientists Identify Tomato Plants` Mechanism for Heat Tolerance
- - FAO Analysis Identifies Opportunities, Gaps and Risks Related to Agrifood Climate Solutions
- - Leaf Architecture and Genome Size Variation of Durio zibethinus L. from Jelebu, Negeri Sembilan, Malaysia
- - Key Eggplant Gene Boosts Resistance to Bacterial Wilt
- - A putative E3 ubiquitin ligase substrate receptor degrades transcription factor SmNAC to enhance bacterial wilt resistance in eggplant
- - Kenya Assures Public of Biosafety Guidelines
- - Leading Breakthroughs: Gene Drives for a Sustainable Agriculture and Biodiversity Conservation
- - Mismatch between lab-generated and field-evolved resistance to transgenic Bt crops in Helicoverpa zea
- - OHRECA fellows reach the finals of the 2024 Transformative Research Challenge with innovative proposal on wild meat
- - Agriculture at COP29: A Vital Conversation for Climate and Food Security
- - CRISPR/Cas knockout of the NADPH oxidase gene OsRbohB reduces ROS overaccumulation and enhances heat stress tolerance in rice
- - Study Finds OsRbohB Knockout Increases Rice Heat Tolerance
- - Myosin-binding protein 13 mediates primary seed dormancy via abscisic acid biosynthesis and signaling in Arabidopsis
- - CRISPR Unlocks Secret to Seed Dormancy
- - UK-Based Start-Up Phytoform`s Gene-Edited Tomato Produces Up to 400% More Fruit in Vertical Farms
- - QTL mapping and genome-wide association analysis reveal genetic loci and candidate gene for resistance to gray leaf spot in tropical and subtropical maize germplasm
- - Researchers Fine Tune the Ability of Agrobacterium to Engineer Plants and Fungi
- - EFSA GMO Panel Concludes GM Maize DP51291 As Safe As Conventional Counterpart
- - OsHRZ1 negatively regulates rice resistant to Magnaporthe oryzae infection by targeting OsVOZ2
- - Study Shows Link Between Iron Signaling and Rice Pathogen Defenses
- - Researchers Publish Updated Genome Sequence of Grass Pea
- - Advances in the evolution research and genetic breeding of peanut
- - Scientists from Vilnius University Reveal a Unique Method to Silence Genes
- - Research Unlocks Secrets of Nitrogen Use in Potatoes
- - A conserved juxtamembrane motif in plant NFR5 receptors is essential for root nodule symbiosis
- - Pakistan Approves GM Soybean Imports
- - Australian OGTR Receives License Application for Commercial Release of GM Mosquitoes
- - Alternative polyadenylation profiles of susceptible and resistant rice (Oryza sativa L.) in response to bacterial leaf blight using RNA-seq
- - International CGE Modeling Training Program in New Delhi
- - A new partnership for climate-sensitive programming in Vietnam: Exploring the links between human mobility, food, and nutrition security in the Mekong Delta
- - Fine mapping of a major co-localized QTL associated with self-incompatibility identified in two F2 populations (broccoli × cauliflower and cauliflower × Chinese kale)
- - UN Climate Change Conference Baku - November 2024
- - Biodiversity, Gender, and Ethnicity Panel: A space for interethnic dialogue and conservation!
- - Artificial selection of two antagonistic E3 ubiquitin ligases finetunes soybean photoperiod adaptation and grain yield
- - Small Island Developing States (SIDS) Solutions Forum 2024 opens in Fiji to share agrifood transformation solutions
- - Unhealthy dietary patterns drive $8 trillion in annual hidden costs of global agrifood systems
- - Genetic dissection of resistance to Phytophthora sojae using genome-wide association and linkage analysis in soybean [Glycine max (L.) Merr.]
- - PhilRice Calls for Reversal of Court Decision on Golden Rice
- - APHIS Issues Regulatory Status Reviews of GM Corn, Almond, and Tomato
- - QTL mapping and BSR-seq revealed loci and candidate genes associated with the sporadic multifoliolate phenotype in soybean (Glycine max)
- - EFSA GMO Panel Releases Statement on the Assessment of GM Soybean MON 87705 × MON 87708 × MON 89788
- - Bioengineers Propose Electro-Agriculture to Produce Food in the Dark and with 88% Less Land
- - Spatiotemporal transcriptomic landscape of rice embryonic cells during seed germination
- - FAO Highlights Role of Genetic Revolution in Addressing Food Security
- - Researchers Reveal Novel Genetic Basis of Pest Resistance to Biotech Crops
- - Knockout of the sugar transporter OsSTP15 enhances grain yield by improving tiller number due to increased sugar content in the shoot base of rice (Oryza sativa L.)
- - European Commission Authorizes Four GE Crops for Import
- - NUS Scientists Produce Lab-Grown Pork with Red Sorghum Grain
- - Genome-wide identification and data mining reveals major-latex protein (MLP) from the PR-10 protein family played defense-related roles against phytopathogenic challenges in cassava (Manihot esculenta Crantz)
- - Gene Editing in Watermelon Confers Resistance Against Zucchini Yellow Mosaic Virus
- - Industry Stakeholders in Scotland Call on Government to Consider Precision Breeding Technology
- - Expression Pattern and Functional Analysis of MebHLH149 Gene in Response to Cassava Bacterial Blight
- - Colombia is 153rd Country to Ratify International Treaty on Plant Genetic Resources for Food and Agriculture
- - CBD Urges Nations to Shift to Emergency Mode to Achieve Biodiversity Targets
- - Anomalous wet summers and rising atmospheric CO2 concentrations increase the CO2 sink in a poorly drained forest on permafrost
- - USDA, EPA, and FDA Launch New Web-Based Tool for Developers of Microbial Biotechnology Products
- - EFSA GMO Panel Finds No Safety Concerns with the Food Enzyme Carboxypeptidase C from GM Aspergillus Niger
- - Genetic loci associated with sorghum drought tolerance in multiple environments and their sensitivity to environmental covariables
- - Researchers Develop Tomato Plants that Contain Full Genetic Material of Both Parent Plants
- - Ban on GM Corn Could Exacerbate Food Insecurity and Job Loss in Mexico
- - Mitosis instead of Meiosis - Researchers breed tomato plants that contain the complete genetic material of both parent plants
- - USDA APHIS Releases Regulatory Status Review for Modified Sweet Orange and Maize
- - Contributions of Biotech Crops to Food Security, Sustainability, and Climate Change Solutions
- - Mechanism of Rice Resistance to Bacterial Leaf Blight via Phytohormones
- - ICRISAT Showcases Innovations in Legume Research at International Conference in Australia
- - ICRISAT and SKUAST Co-Develop First Cold-Tolerant Sorghum in Jammu and Kashmir, India
- - IDD10-NAC079 transcription factor complex regulates sheath blight resistance by inhibiting ethylene signaling in rice
- - Establishing a livestock production and trade alliance to improve market access for small-scale farmers
- - CGIAR Foresight Initiative: New online training materials on economic modeling
- - Studies of genetic diversity and genome-wide association for vitamin C content in lettuce (Lactuca sativa L.) using high-throughput SNP arrays
- - SAPLING assessment shows livestock farmer groups have increased adoption of innovative practices in Vietnam
- - At UN Biodiversity COP16, FAO steers countries to find solutions in agrifood systems
- - Natural alleles of LEAFY and WAPO1 interact to regulate spikelet number per spike in wheat
- - Democratic Republic of the Congo: FAO sounds alarm over persisting high levels of hunger
- - The genetic revolution can support food security, tackle the climate crisis and protect biodiversity
- - Optimization of cassava (Manihot esculenta Crantz) grafting technique to enhance its adoption in cassava cultivation
- - NBSAP Accelerator Partnership paves the way for action
- - Boosting India`s Rice Yields: A data-driven approach to sustainable agriculture
- - Intensive leaf cooling promotes tree survival during a record heatwave
- - Tackling water scarcity through strengthening leadership
- - From commitments to actions: Making Agroecosystem Living Labs work for the Kunming-Montreal Global Biodiversity Framework
- - Genetic loci associated with sorghum drought tolerance in multiple environments and their sensitivity to environmental covariables
- - Bolivia Approves Planting of GM Soybean Intacta
- - USDA APHIS Releases Regulatory Status Review Responses to CoverCress, GCMBNA, Moolec, and MSU
- - Historical Review of Sugarcane Streak Mosaic Virus that Has Recently Emerged in Africa
- - ASEAN-CGIAR presents strategic program at the 46th AMAF PrepSOM
- - Vietnam`s MARD endorses Mechanized Direct Seeding combined with Fertilizer Deep Placement as an advanced technology for rice cultivation
- - A single-nucleotide insertion in Rxp confers durable resistance to bacterial pustule in soybean
- - FAO`s work helps guide priorities and outcomes for G7 and G20 leaders
- - Study Explores Use of Humor in Communicating Gene Editing
- - 16S rRNA metagenomic dataset on endophytic bacterial community of the cashew plant (Anacardium occidentale L.) grown in Dak Lak Province of Vietnam
- - Gene-edited Rice Shows Resistance to Aging
- - CRISPR Enhances Salt Tolerance of Soybeans
- - Prolongation of seed viability and grain quality in rice by editing OsLOX1 using CRISPR/Cas9
- - Gene Editing to Boost Yam Production in Africa
- - Watershed, Evogene, and BGU to Expand Gene Editing Applications to White Leg Shrimp and Red Swamp Crayfish
- - An efficient CRISPR-Cas12a-mediated MicroRNA knockout strategy in plants
- - Researchers Identify Gene Regulating Flowering Time and Leaf Angle in Rapeseed
- - COGEM Releases Advice on Renewal of Import and Processing of GM Cotton T304-40
- - Knockdown of β-conglycinin α` and α subunits alters seed protein composition and improves salt tolerance in soybean
- - Study Finds Cas12a More Effective than Cas9 in Generating Knockout Mutants in MiRNA Genes in Rice
- - Researchers Discover Novel Anti-Stress Molecule in Plants
- - GW3, encoding a member of the P450 subfamily, controls grain width by regulating the GA4 content in spikelets of rice (Oryza sativa L.)
- - MARA Issues List of GM Maize and Soybean Approved for Planting in China
- - Gene Editing to Produce Oats with Improved Nutritional Value and Shelf Life
- - First investigation into the genetic control of meiosis in sugarcane
- - Biofortified Forage, Summer Pearl Millet, and Multi-Trait Cultivars Lead Discussions at Pearl Millet Scientists` Field Day
- - Knowledge and tradition coalesce at the Global-Hub on Indigenous Peoples` Food Systems
- - Refinement of rice blast disease resistance QTLs and gene networks through meta-QTL analysis
- - RTB Breeding Network Formalizes Collaboration to Accelerate Genetic Gains for Smallholder Farmers in Sub-Saharan Africa
- - World Food Day 2024: The critical role of healthy diets for realizing the right to food
- - A receptor for dual ligands governs plant immunity and hormone response and is targeted by a nematode effector
- - Sustainable Homestead Farming for Better Food and Nutrition Security in Bangladesh: Farida Begum`s success story
- - Breaking the cycle of poverty through Mechanized Farming: The Story of Madhuri Mallick
- - Innovations through crop switching happen on the diverse margins of US agriculture
- - The 13th National Seed Congress
- - Vietnam`s 1-Million Hectare Rice Program shows promising results
- - Genome-wide identification of sweet potato U-Box E3 ubiquitin ligases and roles of IbPUB52 in negative regulation of drought stress
- - World Food Forum: Paradise Bank, a forest-themed art exhibition opens at The Rome Botanical Gardens
- - FAO Announces new Food and Agriculture Museum and Network in Rome during audience with Italian President Mattarella
- - Co-localization of quantitative trait loci for pod and kernel traits and development of molecular marker for kernel weight on chromosome Arahy05 in peanut (Arachis hypogaea L.)
- - Women`s network for environmental advocacy and climate resilience: a model for social innovation
- - Building on Bundelkhand Pilot Success: Uttar Pradesh Officials Explore Expansion Avenues
- - Intestinal Lactobacillus murinus-derived small RNAs target porcine polyamine metabolism
- - The Nobel Prize in Chemistry 2024
- - Nobel Prize Honors Scientists Who Discovered MicroRNA
- - Advancing sustainable rice production in the Vietnamese Mekong Delta insights from ecological farming systems in An Giang Province
- - Bangladeshis Demonstrate Positive Stance and Support in Bt Brinjal
- - GM Tomatoes Could Increase Net Income of Lebanese Farmers by USD 50,000 Within Five Years
- - Exploiting DNA methylation in cassava under water deficit for crop improvement
- - Kenya`s Biosafety Authority Prepares to Release More GM Crops
- - Researchers Show New Wheat Varieties with Higher Yields
- - Genomic estimated selection criteria and parental contributions in parent selection increase genetic gain of maternal haploid inducers in maize
- - Gene Editing to Produce Bacterial Blight Resistant Cassava
- - Scientists Reveal Key Genetic Mechanism that Improves Sorghum`s Drought Tolerance
- - Editing of the MeSWEET10a promoter yields bacterial blight resistance in cassava cultivar SC8
- - EU Researchers and Farmers Call on Members of Parliament to Embrace NGTs
- - ISAAA Inc. Provides Collective Learning Space for Product Stewardship Discussions
- - Integrating targeted genetic markers to genotyping-by-sequencing for an ultimate genotyping tool
- - PH Agribusiness Reps Discuss Role of Biotech in Food Security
- - EFSA: Category 1 NGT Plants Equivalent to Conventional Breeding
- - Genomic selection for agronomical phenotypes using genome-wide SNPs and SVs in pearl millet
- - Wageningen Researchers and Partners Develop First Banana Resistant to TR4 and Black Sigatoka
- - Australia`s GM Regulator Approves Field Trial of CSIRO`s GM Canola
- - Understanding public perspectives on genetically engineered Brinjal and the adoption of modern biotechnology in Bangladesh
- - UK Introduces Legislation Supportive of Precision Breeding
- - Responsible Innovation: Ensuring Product Stewardship in Crop Biotech
- - The complete chloroplast genome of Durio zibethinus L. cultivar Ri6 (Helicteroideae, Malvaceae)
- - Vietnam charts new path for sustainable livestock development with CLEANED tool integration
- - SAPLING and Agricultural Service Centre partner to train farmers on sustainable livestock production in Mai Son, Vietnam
- - Cassava Breeding and Cultivation Challenges in Thailand: Past, Present, and Future Perspectives
- - International Seed Federation Calls for Harmonized Regulations on New Breeding Technologies
- - CRISPR Improves Resistance to Bacterial Blight in Rice
- - Enhancing resistance to bacterial blight in rice using CRISPR-based base editing technology
- - Gene Editing Used to Increase Protein in Staple Crops to Alleviate Global Protein Shortage
- - CRISPR Increases Seed Protein Levels in Rice and Soybean
- - A novel SNP within the Rsa10025320 gene is highly associated with hollowness in red-skinned radish fleshy roots
- - UZH Researchers Engineer TnpB into a Compact Gene Editing Tool
- - Responsible Innovation: Ensuring Product Stewardship in Crop Biotech
- - Histopathological Alterations in Nilaparvata lugens (Hemiptera: Delphacidae) after Exposure to Cordyceps javanica
- - Indian Experts Point Out Hurdles in Gene Editing Applications in Crops
- - Research on Plant Stem Cells Shines Light on How Plants Grow Stronger
- - Candidate gene analysis of rice grain shape based on genome-wide association study
- - Kit Makes CRISPR Education Affordable and Accessible to High School Students
- - Engineering Rice Phytobiome Could Lead to Greater Food Security and Combat Climate Change
- - Field performance and nitrous oxide emissions of transgenic nitrogen use efficient rice lines cultivated in tropical paddy fields
- - World Leaders Adopt UN Pact for the Future
- - Nitrogen Use Efficient Rice Shows Promising Field Performance
- - Fine mapping and identification of ERF transcription factor ERF017 as a candidate gene for cold tolerance in pumpkin
- - Inclusive communication campaigns drive the adoption of improved seeds in Uganda
- - Rice phytobiome engineering could lead to greater food security, says IRRI and UC Davis scientists
- - Genome-Wide Analysis of Trehalose-6-Phosphate Phosphatase Gene Family and Their Expression Profiles in Response to Abiotic Stress in Groundnut
- - FAO leads new initiative and reinforces its commitment to accelerate gender equality efforts in agrifood systems
- - Can crop breeding fight food loss and waste?
- - Comparative analysis of infected cassava root transcriptomics reveals candidate genes for root rot disease resistance
- - Fostering collaboration for healthier diets in Southeast Asia
- - Eating wild meat: The rewards are as big as the risks
- - SlJMJ14, identified via QTL seq and fine mapping, controls flowering time in tomatoes
- - CGIAR innovation joint village land use planning making policy gains
- - Join CGIAR Gender Equality Initiative, HER+ for a High-Level Dialogue on Gender and Climate Change in Nigeria, Abuja
- - Rapid growth and the evolution of complete metamorphosis in insects
- - FAO opens groundbreaking conference aimed at tackling the threat of animal diseases
- - UNGA79: FAO welcomes adoption of UN Pact for the Future
- - GROWTH REGULATING FACTOR 7-mediated arbutin metabolism enhances rice salt tolerance
- - The potential of bundle innovation in closing gender yield gaps
- - Goats with Aprons? A traditional anti-mating innovation used by Maasai communities in Tanzania
- - Mapping and transcriptomic profiling reveal that the KNAT6 gene is involved in the dark green peel colour of mature pumpkin fruit (Cucurbita maxima L.)
- - Scientists Use CRISPR to Modify Euglena for Biofuel Production
- - Century-Old Experiment and New Research Identify Genes that Help Barley`s Adaptability
- - Genome-Wide Analysis of the Auxin/Indoleacetic Acid (Aux/IAA) Gene Family in Autopolyploid Sugarcane (Saccharum spontaneum)
- - COGEM Releases Advice on Renewal of Import and Processing of GM Cotton MON15985
- - Rice Gene Confers Water-deficit Stress Tolerance in Arabidopsis
- - CRISPR/Cas9-mediated multiplex gene editing of gamma and omega gliadins, paving the way for gliadin-free wheat
- - FSANZ Opens Public Comment Period for GM Sugar Beet
- - Health and Environmental Concerns Influence Consumers` GM Food Consumption
- - Flexibility of parental-like or maternal-like gene expression under diverse environments contributes to combined drought avoidance and drought tolerance in a water-saving and drought-resistance rice hybrid
- - Gene Editing to Produce Gluten-Free Wheat
- - Researchers Develop Biofortified Golden Lettuce with 30 Times More Beta Carotene
- - Redesigning soybean with improved oil and meal traits
- - Webinar highlights innovations for climate-resilient agriculture in Asian Mega-Deltas
- - Transforming Soil Health in West Africa: A New Fertilizer and Soil Health Hub Takes Root in West Africa
- - Map-based cloning of LPD, a major gene positively regulates leaf prickle development in eggplant
- - Harmonization of Gene Editing Regulations Vital in Bringing Products to Market
- - Researchers Investigate the Vegetative Growth of Tomato Through Gene Editing
- - The SINA1-BSD1 Module Regulates Vegetative Growth Involving Gibberellin Biosynthesis in Tomato
- - Effective CRISPR RNAs Selection Leads to Less Off-target Edits
- - Gene Editing Produces Blast-Resistant Rice
- - Sugar signaling modulates SHOOT MERISTEMLESS expression and meristem function in Arabidopsis
- - Study Finds Agriculture Accelerated Human Genome Evolution to Capture Energy from Starchy Foods
- - Global Study Identifies Practices for Climate-Resilient Agriculture
- - Current status of molecular rice breeding for durable and broad-spectrum resistance to major diseases and insect pests
- - COGEM Releases Advice on Renewal of Import and Processing of GM Cotton MON15985
- - EFSA Issues 2022 Post-market Environmental Monitoring Report on the Cultivation of GM Maize MON 810
- - ZmABF4-ZmVIL2/ZmFIP37 module enhances drought tolerance in maize seedlings
- - ASCA7 Provides Platform for Learning, Collaboration, and Empowerment
- - CABBI Engineers Bioenergy Crops for Improved Water Efficiency
- - OsNAC120 balances plant growth and drought tolerance by integrating GA and ABA signaling in rice
- - Chinese Researchers Highlight the Need for Equitable Access to CRISPR Technologies
- - ASEAN Nations Tackle Gene Editing Applications and Science-based Regulations
- - Phenoxyacetic acid enhances nodulation symbiosis during the rapid growth stage of soybean
- - Bilateral meeting with H.E. Shavkat Mirziyoyev, President of the Republic of Uzbekistan
- - Private-public partnerships are key to agrifood systems transformation
- - Peptide REF1 is a local wound signal promoting plant regeneration
- - COGEM Concludes Import and Processing of GM Soybean MON 87708 Does not Pose an Environmental Risk in The Netherlands
- - Researchers Release First Complete Telomere-to-Telomere Genome of Lettuce
- - Allelic variation in rice blast resistance: a pathway to sustainable disease management
- - New Gene Editing Technique Allows Subtle Modifications to Spread Through Wild Populations
- - FAO Pushes for Innovation for a Sustainable Future of Forests
- - Mapping rust resistance in European winter wheat: many QTLs for yellow rust resistance, but only a few well characterized genes for stem rust resistance
- - Gene-edited Dairy Goats Exhibit Enhanced Resistance to Mastitis
- - USDA APHIS Releases Regulatory Status Review on HB4 Wheat
- - Multiomics of a rice population identifies genes and genomic regions that bestow low glycemic index and high protein content
- - IRRI Researchers Identify Genes and Markers for Low Glycemic Index and High Protein in Rice
- - Experts Develop Tiny Tomatoes for Astronauts to Grow in Space
- - Knockout of OsWOX13 Moderately Delays Flowering in Rice under Natural Long-Day Conditions
- - OsWOX13 Controls Rice Flowering and Drought Escape
- - CRISPR/CasRx-mediated resistance to Soybean mosaic virus in soybean
- - CRISPR Improves Resistance of Soybeans to Soybean Mosaic Virus
- - Genome-wide association study of fiber quality traits in US upland cotton (Gossypium hirsutum L.)
- - Chinese Cherry Genome Reveals Genes for Firmer Fruit Texture
- - A review and outlook on expression of animal proteins in plants
- - Researchers Review the Impact of Animal Proteins in Plants
- - Key transcription factors regulate fruit ripening and metabolite accumulation in tomato
- - ISAAA Inc. Provides Information on the Recent Developments in Gene Editing
- - Genome-wide association study and selective sweep analysis uncover candidate genes controlling curd branch length in cauliflower
- - Last Chance to Join ASCA7 in Bangkok
- - Genome editing prospects for heat stress tolerance in cereal crops
- - Transcriptome analysis of differentially expressed genes in rice seedling leaves under different nitrate treatments on resistance to bacterial leaf blight
- - Philippine Agriculture Delegation Visits ICRISAT-led Project Sites in India
- - Detection and Evaluation of Blast Resistance Genes in Backbone Indica Rice Varieties from South China
- - A key ABA biosynthetic gene OsNCED3 is a positive regulator in resistance to Nilaparvata lugens in Oryza sativa
- - How drones are used to model river flows on a regional scale
- - Decline in carbon emission intensity of global agriculture has stagnated recently
- - Change of training approach improves farmers` reception to SAPLING innovations in Vietnam
- - Integrating RTM-GWAS and meta QTL data revealed genomic regions and candidate genes associated with the first fruit branch node and its height in upland cotton
- - Review Shows Advancements in Gene Editing of Sorghum for Crop Improvement
- - Deciphering salt stress responses in Solanum pimpinellifolium through high-throughput phenotyping
- - Researchers Analyze Salt Stress Tolerance of Wild Tomatoes
- - Consumer Acceptance of Folate-Biofortified GM Lettuce in Brazil
- - Morphological, anatomical, and transcriptomics analysis reveals the regulatory mechanisms of cassava plant height development
- - EFSA GMO Panel Concludes GM Maize MON 95275 As Safe As Conventional Counterpart
- - Clemson University Researchers Uncover Roots of Climate-Resilient Cotton
- - Distribution, characterization and chemical management of noxious shrub weed (Dichapetalum stuhlmannii Engl) in cashew in southeastern Tanzania
- - Researchers Provide Recommendations to Ensure Accessibility and Affordability of Foods Derived from Biotech
- - An artificially evolved gene for herbicide-resistant rice breeding
- - Cassava molecular genetics and genomics for enhanced resistance to diseases and pests
- - Scientists Use CRISPR to Enhance Aroma Quality of Apple
- - Gene Editing of Grain Size Genes Leads to Better Rice Appearance and Yield
- - Exploration and genetic analyses of canopy leaf pigmentation changes in soybean (Glycine max L.): unveiling a novel phenotype
- - EFSA GMO Panel Concludes GM Maize DP910521 As Safe As Conventional Counterpart
- - m6A modification plays an integral role in mRNA stability and translation during pattern-triggered immunity
- - Biosensor Reveals Gibberellin`s Critical Role in Legume Nitrogen-Fixation
- - Genetic diversity and genome-wide association study of partial resistance to Sclerotinia stem rot in a Canadian soybean germplasm panel
- - Transcriptome Analysis of Early Lateral Root Formation in Tomato
- - FAO Food Price Index broadly unchanged in July
- - Gaza: FAO delivers more than 2 400 veterinary kits to protect livestock and sustain livelihoods
- - Transcriptome analysis reveals genes associated with late blight resistance in potato
- - The Nation: Why we need to invest in sustainable food systems
- - Ubiquitin-specific protease UBP14 stabilizes HY5 by deubiquitination to promote photomorphogenesis in Arabidopsis thaliana
- - CSSP-SABRAO awards Jauhar Ali prestigious Sant S. Virmani Hybrid Rice prize
- - Gut bacteria are essential for development of an invasive bark beetle by regulating glucose transport
- - Breeding and management of major resistance genes to stem canker/blackleg in Brassica crops
- - Famine conditions in parts of Sudan: FAO urges at scale life-saving assistance to boost local food production
- - Durian fruit pulp extract enhances intracellular glutathione levels, mitigating oxidative stress and inflammation for neuroprotection
- - STEP`s remarkable impact cultivating future agribusiness leaders
- - Mining for QTL controlling maize low-phosphorus response genes combined with deep resequencing of RIL parental genomes and in silico GWAS analysis
- - Mapping of a novel locus Ra conferring extreme resistance against potato virus A in cultivated potato (Solanum tuberosum L.)
- - Biosynthesis of Scopoletin in Sweet Potato Confers Resistance against Fusarium oxysporum
- - Gene Editing Produces Pigs Resistant to Influenza A Virus
- - Genetically Engineered Black Flies to Reduce Waste and Keep it Out of Landfills
- - Analysis of Potato Physiological and Molecular Adaptation in Response to Different Water and Nitrogen Combined Regimes
- - 7th Asian Short Course on Agribiotechnology, Biosafety Regulation, and Communication ISAAA
- - Prediction of resistance, virulence, and host-by-pathogen interactions using dual-genome prediction models
- - Bud shapes dictate tiller–rhizome transition in African perennial rice (Oryza longistaminata)
- - CRISPR Promotes Maturation in Soybeans
- - Groundcherry Gets Genetic Upgrades Through CRISPR
- - Engineering high amylose and resistant starch in maize by CRISPR/Cas9-mediated editing of starch branching enzymes
- - Researchers Present Improved Direct PCR-based Technology for GE Plants Screening
- - Root system architecture, physiological analysis and dynamic transcriptomics unravel the drought-responsive traits in rice genotypes
Independence Award
- First Rank - Second Rank - Third Rank
Labour Award
- First Rank - Second Rank -Third Rank
National Award
- Study on food stuff for animal(2005)
- Study on rice breeding for export and domestic consumption(2005)
VIFOTEC Award
- Hybrid Maize by Single Cross V2002 (2003)
- Tomato Grafting to Manage Ralstonia Disease(2005)
- Cassava variety KM140(2010)
-
Department of Biotechnology
https://sites.google.com/site/cadcnshias/ -
Dalat center
http://pvfcdalat.vn -
Hung Loc Agricultural Research Center
http://harc-ias.vn/

 Curently online :
8 Curently online :
8 |
|
 Total visitors :
8923581 Total visitors :
8923581 |
|
|
Genome-Wide Association and Gene Co-expression Network Analyses Reveal Complex Genetics of Resistance to Goss`s Wilt of Maize.
Thursday, 2019/08/15 | 07:09:56
|
||||||||||||||||||||||||||||||||||||||||
|
Singh A, Li G, Brohammer AB, Jarquin D, Hirsch CN, Alfano JR, Lorenz AJ. G3 (Bethesda). 2019 Jul 30. pii: g3.400347.2019. doi: 10.1534/g3.119.400347. AbstractGoss's bacterial wilt and leaf blight is a disease of maize caused by the gram positive bacterium Clavibacter michiganensis subsp. nebraskensis (Cmn). First discovered in Nebraska, Goss's wilt has now spread to major maize growing states in the United States and three provinces in Canada. Previous studies conducted using elite maize inbred lines and their hybrids have shown that resistance to Goss's wilt is a quantitative trait. The objective of this study was to further our understanding of the genetic basis of resistance to Goss's wilt by using a combined approach of genome-wide association mapping and gene co-expression network analysis. Genome-wide association analysis was accomplished using a diversity panel consisting of 555 maize inbred lines and a set of 450 recombinant inbred lines (RILs) from three bi-parental mapping populations, providing the most comprehensive screening of Goss's wilt resistance to date. Three SNPs in the diversity panel and 10 SNPs in the combined dataset, including the diversity panel and RILs, were found to be significantly associated with Goss's wilt resistance. Each significant SNP explained 1% to 5% of the phenotypic variation for Goss's wilt (total of 8 - 11%). To augment the results of genome-wide association mapping and help identify candidate genes, a time course RNA sequencing experiment was conducted using resistant (N551) and susceptible (B14A) maize inbred lines. Gene co-expression network analysis of this time course experiment identified one module of 141 correlated genes that showed differential regulation in response to Cmn inoculations in both resistant and susceptible lines. SNPs inside and flanking these genes explained 13.3% of the phenotypic variation. Among 1,000 random samples of genes, only 8% of samples explained more phenotypic variance for Goss's wilt resistance than those implicated by the co-expression network analysis. While a statistically significant enrichment was not observed (P < 0.05), these results suggest a possible role for these genes in quantitative resistance at the field level and warrant more research on combining gene co-expression network analysis with quantitative genetic analyses to dissect complex disease resistance traits. The results of the GWAS and co-expression analysis both support the complex nature of resistance to this important disease of maize.
See https://www.ncbi.nlm.nih.gov/pubmed/31362973
Figure 3: Comparison of physical positions of QTL detected in bi-parental linkage 968mapping and GWAS. Bi-parental linkage mapping was conducted by Singh et al. 969(2016),and significant SNPs from GWAS in the combined dataset co-located on970chromosomes 1, 2, and 5. The x-axis of each plot represents the physical positionof each 971SNP,and the y-axis displays the negative log10of p-valuesfor eachSNPincluded in the 972GWAS. Gray colored solid points represent all SNPs used in GWAS. Significant SNPs in 973the GWASare indicated by dark green dots, and 2-LOD support interval ofQTL detected 974by Singh et al. (2016) are shown by the red or blue windows. |
||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||
|
[ Other News ]___________________________________________________
|
(44).png "Genome-Wide Association and Gene Co-expression Network Analyses Reveal Complex Genetics of Resistance to Goss`s Wilt of Maize.")